CHAPTER 46 Neurodegeneration: Cerebellum and Brain Stem
The focus of this chapter is on the neurodegenerative diseases that primarily affect the brain stem and cerebellum, although they may also involve other areas. In these conditions, imaging is used to exclude other diseases that may mimic neurodegeneration, such as demyelination, vasculopathies, normal pressure hydrocephalus, and tumor, and to plan stereotactic surgery to control the symptoms of Parkinson’s disease.1 Advances in transcranial ultrasonography, MRI, functional imaging with positron emission tomography (PET), and single photon emission computed tomography (SPECT) offer hope that the diverse forms of neurodegeneration may soon be assessed directly. CT contributes little to the evaluation of neurodegenerative diseases, so it is not discussed further.
Neurodegenerative diseases become symptomatic in the later stages of adult life, so it is important to be able to distinguish these diseases from the effects of normal aging on brain volume and signal intensity. Age-related volume changes are usually diffuse with no regional predilection. Age-related changes in signal intensity largely reflect the deposition of metals, especially iron, within the brain. In the normal human brain, iron deposition is symmetric and geographic, affecting primarily the globus pallidus, the striatum, the substantia nigra (especially the pars reticulata), and the dentate nucleus of cerebellum. Quantitative techniques show that the highest concentrations of iron are found in the globus pallidus, red nucleus, substantia nigra, putamen, dentate, and caudate nuclei.2 T2-weighted (T2W) MR images at 1.5 Tesla show normal, age-related hypointensities within the globus pallidus, substantia nigra pars reticulata, red nucleus, dentate, putamen, and the subcortical U fibers.3 Postmortem studies with Perls’ stain for ferric iron reveal that the areas of T2 hypointensity correlate with regions of intense Perls’ staining. However, there is an imperfect match between the distribution of iron and the hypointensity on T2W images. Specifically, the putamen and caudate nucleus are less hypointense than would be expected from Perls’ stain data, whereas the corpus callosum, other commissures, and internal capsule appear more hypointense than would be expected from Perls’ stain data. However, MRI studies of the effect of age on T2 signal intensity within the deep gray nuclei are in agreement with the pathologic studies. There is no iron deposition at birth. Iron becomes deposited, and causes low T2 signal intensity, in order, first within the globus pallidus, then the red nucleus and substantia nigra, and then the dentate nuclei of the cerebellum.4 By age 25 years, these structures are all hypointense relative to cortical gray matter. During adult life the signal intensity within the red nucleus, dentate, and substantia nigra remain relatively stable. The pallidum becomes progressively more hypointense. The putamen becomes hypointense in the eighth and ninth decades. The thalamus and the caudate nucleus are not hypointense in healthy populations.5
PARKINSON’S DISEASE
Clinical Presentation
Bradykinesia may manifest as slowness in manual tasks, difficulty in buttoning buttons or using a keyboard, a reduction in arm swing while walking, mask-like facial expression, and micrographia. The Parkinson tremor is classically a 4- to 6-Hz resting tremor. The gait is shuffling and festinant, leading to postural instability and falls. Nonmotor features of PD include hyposmia and disorders of rapid-eye-movement sleep (which may substantially precede the motor symptoms). Neuropsychiatric features include depression and dementia. Autonomic features include urinary incontinence, disorders of sweating, orthostatic hypotension, and erectile dysfunction. Disorders of the gastrointestinal tract (constipation) and sensory disturbances may also be present. Medical treatment is required within the first few years of disease. Present symptomatic treatments aim to correct the dopamine deficit found in PD. More than 90% of patients show good initial clinical response to levodopa, but the drug is associated with later motor complications, including a reduction in the duration of drug action, leading to a “wearing off” effect, choreiform dyskinesias, and unpredictable “off periods.” These complications require use of other medications to control them by stimulating dopaminergic receptors (dopamine agonists), reducing the rate of dopamine breakdown (COMT inhibitors, MOAB inhibitors), and other mechanisms (amantadine). With these adjunctive therapies, it is often possible to control PD symptoms adequately, without significant adverse reactions for many years. Some patients require infusions (duodopa enteral infusions, apomorphine subcutaneous infusions) and/or deep brain surgery to the pallidum and subthalamic nucleus (lesioning or deep electrode stimulation). Disease progression may be associated with postural instability, impaired speech, and cognitive decline, and by 15 years after disease onset these may become the most disabling symptoms. By 20 years after onset 74% of PD patients have died and 84% of those living suffer dementia.
Pathology
Two-Hit Hypothesis
With normal aging, pigmented neurons of the SNc are lost at a rate of 4.7% per decade. In PD, 45% of SNc cells are lost in the first decade of disease, especially from the lateral ventral tier. On gross inspection, therefore, the SNc and the locus ceruleus are visibly pale to the naked eye in PD (Fig. 46-1). This cell loss leads to selective loss of dopaminergic innervation of the striatum and explains the pattern of reduced presynaptic nigrostriatal uptake seen in PET and SPECT studies (see later). Pathologic correlation remains important, because postmortem series have shown that up to 10% of those clinically diagnosed with PD during life may turn out to have an alternative diagnosis.
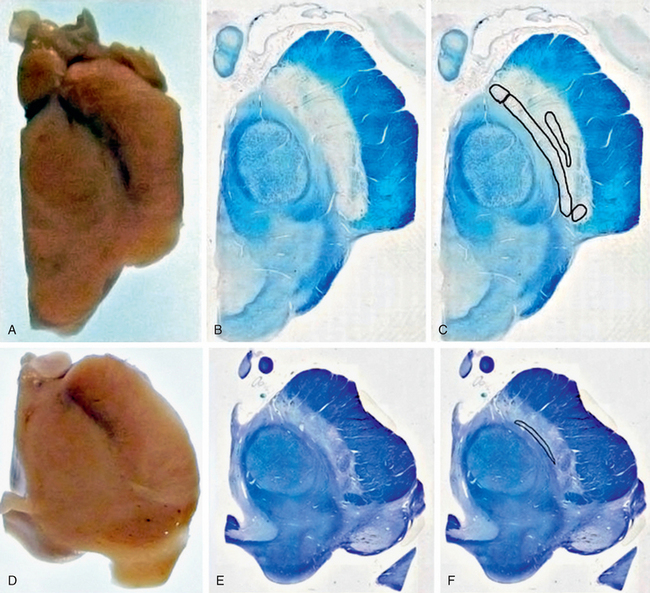
FIGURE 46-1 The substantia nigra. A, Macroscopic image showing the black substance (literal translation of substantia nigra) in control subject. B, Luxol fast blue stain for myelin. C, Clusters of neurons containing neuromelanin pigment. D, Macroscopic image in Parkinson’s disease showing medial preservation but lateral loss of pigment. E, Luxol fast blue stain for myelin. F, Location of remaining pigmented neurons of the SNc.
(Photography courtesy of Susan Stoneham, histological stains courtesy of Kate Strand, both of Queen Square Brain Bank for Neurological Disorders, UCL Institute of Neurology, London.)
The pathologic hallmarks of PD are round eosinophilic intracytoplasmic inclusions (designated Lewy bodies) and dystrophic neurites (designated Lewy neurites). Lewy bodies contain the protein α-synuclein (see Fig. 46-1).
Imaging
Ultrasonography
Transcranial B-mode ultrasonography plays a significant role in the neuroimaging diagnosis of PD and its mimics. Transcranial ultrasonography may be employed successfully in approximately 90% of PD patients, although 10% of patients have temporal acoustic bone windows too small to permit study.6 In PD, transcranial ultrasonography characteristically shows an enlarged area of hyperechogenicity in the substantia nigra (Fig. 46-2). In normal controls, the area of substantia nigra hyperechogenicity measures about 0.12 cm2. Using a cutoff value of 0.19 to 0.2 cm2 to define an area of echogenicity as abnormal (1 standard deviation above the mean), transcranial ultrasonography successfully identifies approximately 90% of patients clinically classified as having PD but also 9% of healthy controls. Increasing the area value to 0.24 cm2 (2 standard deviations) increases the specificity for PD but reduces the sensitivity. Notably, the healthy controls who exhibit substantia nigra hyperechogenicity do show abnormalities on functional imaging, suggesting that a positive ultrasonographic sign may indicate a real underlying pathologic process in the substantia nigra. In support of that, hyperechogenicity in the substantia nigra is found more often in cohorts of older controls (46% of 86- to 95-year-old control patients) and is associated in them with subtle extrapyramidal motor deficits.
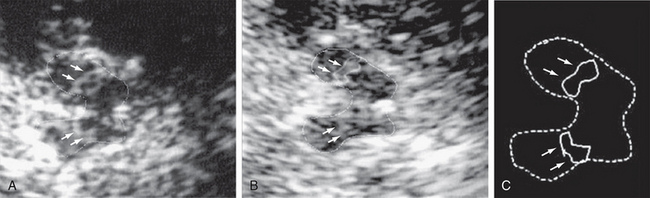
FIGURE 46-2 Transcranial ultrasound in Parkinson’s disease. A, Hypoechogenic mesencephalon (outlined) and hyperechogenic substantia nigra (arrows). B, Second examination of the substantia nigra (outlined) shows no change in area (0.23 cm2). C, Schematic representation of ultrasound image.
(From Berg D, Merz B, Reiners K, et al. Five-year follow-up study of hyperechogenicity of the substantia nigra in Parkinson’s disease. Mov Disord 2005; 20:383-385.)
Other conditions are associated with increased echogenicity in the midbrain. Nigral midbrain hyperechogenicity is found in patients with impaired nigrostriatal function unmasked by neuroleptics and in 16% of patients with essential tremor. In one transcranial ultrasound study of depressed patients, 40% had substantia nigra hyperintensity plus hypoechogenicity in the midbrain raphe nuclei. Ultrasonography can also detect differences in substantia nigra echogenicity in PD versus “atypical parkinsonian syndromes” (see later). A prospective blinded study reported a sensitivity of 90.7%, specificity of 82.4%, positive predictive value (PPV) of 92.9%, and a negative predictive value (NPV) of 77.8% for identifying patients with PD in a mixed cohort of patients that also included individuals with atypical parkinsonian syndromes.6
The pathologic correlate of increased substantia nigra echogenicity is not clear. It could be related to iron deposition, to H- and L-ferritin, and (inversely) to neuromelanin.7 However, hyperechogenicity is not seen in atypical parkinsonian syndromes or other disorders with increased midbrain iron. Furthermore, the globus pallidus is not hyperechogenic despite having a high iron content, so another process is likely to cause the ultrasound echoes.
MRI
On axial T2W MR images of the midbrain, the substantia nigra typically appears as a region of two different signal intensities: (1) a ventrally situated curved band of low signal intensity corresponding to the substantia nigra pars reticularis (SNr) and (2) a dorsally situated curved band of high signal intensity corresponding to the substantia nigra pars compacta (SNc)8 (Fig. 46-3). The low signal red nucleus lies dorsomedial to the pars compacta. In PD, the high signal band of the SNc appears to decrease in breadth, owing to loss of dopaminergic melanized neurons in this region and/or to increased iron deposition (and corresponding signal loss) in the surrounding structures. The distance between the SNr and the red nucleus may be reduced and the hypointensity of the SNr may appear “smudged.” PD patients may show regions of hyperintensity within the lateral SNr (i.e., zones with “restoration of the normal signal intensity”). The thickness of the high signal pars compacta shows inverse correlation with the clinical severity of PD.9 Overall, these changes in the substantia nigra are inconsistent and difficult to reproduce on present 1.5-T scanners. The values measured significantly overlap with controls and do not correlate well with measures of disease severity. Recently, 3-T multiple gradient-echo pulse sequences designed for rapid single-scan mapping of the proton transverse relaxation rate R2* have successfully demonstrated changes in the lateral portion of SNc in PD, corresponding to the known topography of PD pathology.10 In the future, diverse MR techniques such as multiecho T2 and T2* at 3 T, partially refocused interleaved multiple echo (PRIME), and field-dependent increase in R2 (FDRI) may be used to measure the increase in iron deposition in the substantia nigra in PD.
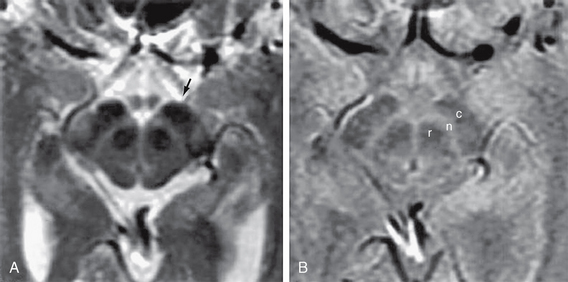
FIGURE 46-3 A, Axial T2W MR image of the midbrain showing the red nuclei and substantia nigra. Arrow indicates the T2 hypointense region corresponding to the SNr. B, Axial proton-density–weighted MR image of the midbrain. c, crus cerebri; n, substantia nigra; r, red nucleus.
(From Oikawa H, Sasaki M, Tamakawa Y, et al. The substantia nigra in Parkinson disease: proton density–weighted spin-echo and fast short inversion time inversion-recovery MR findings. AJNR Am J Neuroradiol 2002; 23:1747-1756.)
Inversion Recovery
On proton density and fast short tau inversion recovery (fast-STIR) images the substantia nigra appears as a tilted band-like structure of hyperintense gray matter.11 This structure corresponds to the substantia nigra described in anatomic atlases but is different from the structure shown on T2W MR images. The region of hypointensity shown on MRI overlaps with only the most superior and ventral portion of the substantia nigra shown on fast STIR.11 Double inversion recovery MR acquisitions that use the ratio of gray matter suppressed to white matter suppressed are sensitive to the neuropathologic changes in the substantia nigra.12 These images show progressive pathologic changes in the substantia nigra with the same lateral to medial and anterior to posterior progression seen in pathologic specimens of PD. However, this technique has also proven difficult to reproduce.
Magnetic Resonance Spectroscopy
Magnetic resonance spectroscopy (MRS) has the potential to detect early pathophysiologic changes before structural abnormalities become evident on conventional MRI. Most reports analyze the spectra of protons (1H-MRS); a few analyze phosphorus (31P-MRS). Proton MRS detects the metabolites N-acetylaspartate (NAA), myoinositol, choline (Cho), and creatine (Cr). Creatine is often used as a reference metabolite and is assumed to be relatively constant (Table 46-1).
TABLE 46-1 Metabolites Commonly Cited in the Literature using 1H-MRS
| Metabolite | Location/Role |
|---|---|
| iV-acetyl-aspartate (NAA) | Amino acid present principally in neurons and thus a marker of neuronal integrity |
| Choline (Cho) | Choline-containing metabolites are involved in phospholipid membrane synthesis |
| Creatine (Cr) | Includes phosphocreatine; relatively unaffected by disease and thus the internal reference standard |
| Others | Includes myoinositol, lactate, and neurotransmitters (GABA, glutamate), etc. |
GABA, γ-aminobutyric acid.
The MRS literature is constrained by methodologic differences among the studies, including the use of small patient groups, differences in disease severity and duration at the time of study, different echo and relaxation times, large voxel sizes leading to partial volume effects, varying pulse sequences, and different field strengths of the magnets. Advances in technology including higher field strengths, improved shimming, and broad intrinsic line widths are first providing quality spectra from the affected region of the brain. To date, most studies report ratios of metabolites, rather than concentrations of metabolites, owing to the difficulties of measuring absolute metabolite values. Single-voxel studies of the lentiform nucleus have generally shown no changes in NAA/Cr ratios in PD. Some studies report reduced NAA/Cr ratios in more-developed PD with motor fluctuations, but spectral results have been inconsistent even within the same group using the same methodology. A recent feasibility study of single-voxel STEAM 1H-MRS (short-echo stimulated echo acquisition mode proton MRS) at 4 T shows that the substantia nigra contralateral to the clinically affected side exhibits an increased γ-aminobutyric acid (GABA)/glutamine ratio when compared with the cortex. This is in accord with pathologic studies.13 Higher field strength and smaller voxel sizes may increase the sensitivity of 1H-MRS to pathologic changes in the future.
Special Procedures
Functional Imaging
The degeneration of the nigrostriatal projection can be mapped by radiotracers using positron emission tomography (PET) or single photon emission computed tomography (SPECT). Radiotracers have been developed to target both the presynaptic and the postsynaptic nigrostriatal systems14–17 (Table 46-2). Although PET remains the gold standard, SPECT has practical advantages over PET, such as the longer half-life of SPECT ligands, which eliminates the need for an on-site cyclotron. Presynaptic targets include (1) the dopamine transporter (DAT), (2) the vesicular monoamine transporter-2 (VMAT-2) that packages dopamine into synaptic vesicles, and (3) aromatic amino acid decarboxylase (AAAD), which converts exogenous levodopa and endogenous tyramine to dopamine. Postsynaptic ligands target the dopamine D2 receptor. Another ligand is directed against activated microglia. Regional metabolic rate can be measured using radiolabeled oxygen or glucose.

Presynaptic Tracers
The projections of the nigrostriatal tract are arranged topographically. PD preferentially involves the ventrolateral tier of the substantia nigra, which projects to the dorsolateral striatum (putamen) and shows relative sparing of the ventral tegmental area, which projects to the ventromedial striatum (caudate nucleus). Therefore, patients with PD show greater loss of dopaminergic innervation in the putamen than in the caudate nucleus (Fig. 46-4). Typical PET and SPECT findings in PD are (1) bilateral reduction in striatal uptake of presynaptic tracers that is greatest on the side contralateral to the most severe symptoms and (2) more severe loss in the putamen than in the caudate nucleus. The measured decline in presynaptic nigrostriatal function correlates with clinical measures of disease progression. For example, early asymptomatic disease manifests as reduced uptake of 18F-dopa on the ipsilateral asymptomatic side in patients with clinical hemiparkinsonism. In families with a history of PD, radionuclide studies show reduced uptake of presynaptic nigrostriatal tracers in those members who subsequently go on to develop PD symptoms and in the asymptomatic monozygotic and dizygotic twins of patients with PD. Functional imaging successfully discriminates PD from essential tremor, wherein presynaptic nigrostriatal dysfunction is unaffected. They distinguish PD from vascular parkinsonism, wherein 123I-β-CIT SPECT is normal or reduced only in accord with the vascular changes seen on MRI or CT. A vascular etiology of disease is suggested by finding reduced uptake on 123I-β-SPECT and MR evidence of a discrete vascular infarct in the striatum. Demonstration of a normal radiotracer scan, indicating no presynaptic dopaminergic dysfunction, is not compatible with PD or other neurodegenerative disease affecting the nigrostriatal tract. In the correct clinical context, such findings point more toward essential tremor, psychogenic parkinsonism, drug-induced parkinsonism, or vascular parkinsonism.18
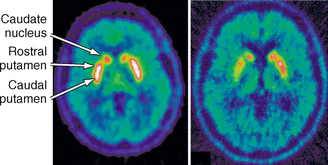
FIGURE 46-4 18F-Dopa PET images of the striatum in a healthy control (left) and a patient with Parkinson’s disease (right) showing asymmetric loss of tracer uptake more severe in the caudal putamen than the rostral putamen or caudate nucleus.
(From Piccini P, Whone A. Functional brain imaging in the differential diagnosis of Parkinson’s disease. Lancet Neurol 2004; 3:284-290.)
Postsynaptic Tracers
Studies with 18F-dopa PET reliably discriminate between controls and parkinsonian illnesses but are less reliable for distinguishing among PD, PSP, and MSA. Patients with atypical parkinsonism, particularly PSP, show a tendency toward lower uptake of tracer in the caudate nucleus than is seen in PD and MSA.19 However, measurements of postsynaptic abnormalities in the striatum using ligands to D2 receptors and fluorodeoxyglucose (FDG)-labeled PET to detect metabolic abnormalities reveal postsynaptic abnormalities in the putamen in MSA (evident as reduction in raclopride binding on FDG-PET), which correlate with the severity of the illness clinically.20 Putaminal binding of raclopride is increased early in PD and subsequently falls, but not as much as it does in PSP and MSA. However, both PSP and MSA have similar postsynaptic abnormalities, so these two conditions are not distinguished by raclopride-binding studies.
Functional Imaging in Preclinical Disease and as a Marker of Disease Progression
In the REAL-PET trial of the dopamine agonist ropinirole versus levodopa, ropinirole significantly slowed the decline in putaminal 18F-dopa uptake by 34%, although the patients on levodopa were clinically better off at 2 years.21 The ELLDOPA trial of levodopa versus placebo showed that, at 9 months, higher doses of levodopa are associated with better clinical outcomes but have increased rates of adverse events and increased nigrostriatal deficiency (manifest as greater 123I-β-CIT SPECT loss).22 Interestingly, significant proportions of the PD subjects in these studies had normal PET or SPECT scans, both at initiation into the studies and at follow up. This group has been termed SWEDD (scans without evidence of dopaminergic deficit) and are unlikely to have PD.
Cardiac MIBG
Static planar imaging is performed 2 to 4 hours after injection of 123I-MIBG, and a ratio of heart to mediastinal (H/M ratio) values is calculated (Fig. 46-5). This ratio compensates for interindividual variation and technical differences between sites and machines. The H/M ratio is lower in PD than MSA, PSP, and controls. All PD patients, including those with less than 2-year disease duration and without autonomic features, can be distinguished from MSA patients. All MSA patients have measurements that fall within normative value ranges, and findings are independent of autonomic function and disease duration.23 Thus, patients with PD may have earlier cardiac autonomic impairment than is clinically evident (usually this is clinical manifest some years into the disease), although patients with greater autonomic dysfunction may have smaller H/M ratios.24 However, there may be cardiac sympathetic degeneration in MSA, particularly if there are concomitant Lewy bodies, and putaminal measurements of the diffusion tensor may be better at discriminating PD and MSA-P.25
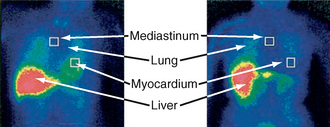
FIGURE 46-5 123I-MIBG scintigraphy in multiple system atrophy and dysautonomic Parkinson’s disease. Anterior view at 2 hours after injection, with overlaid regions of interest placed in order to calculate the heart to mediastinal ratio (H/R ratio).
(From Courbon F, Brefel-Courbon C, Thalamas C, et al: Cardiac MIBG scintigraphy is a sensitive tool for detecting cardiac sympathetic denervation in Parkinson’s disease. Mov Disord 2003; 18:890-897.)
PROGRESSIVE SUPRANUCLEAR PALSY
Clinical Presentation
1. Richardson’s syndrome (RS), classic PSP, accounts for approximately 54% of PSP documented at postmortem. RS presents as postural instability with early falls; a distinctive growling dysarthria; supranuclear gaze palsy; parkinsonian features of bradyphrenia, symmetric bradykinesia, and rigidity that do not respond well to levodopa. They also show frontal type dementia with apathy, reduced verbal fluency, utilization or imitation behavior, and frontal release signs.
2. PSP-Parkinsonism (PSP-P) accounts for approximately 32% of PSP documented at postmortem. PSP-P presents as an asymmetric syndrome with tremor that is difficult to distinguish from PD. The tremor is moderately responsive to levodopa.
3. Approximately 14% of PSP patients present as a clinically heterogeneous group that cannot be categorized into RS or PSP-P.
Imaging
Ultrasonography
PSP does not show the characteristic hyperechogenicity of the substantia nigra seen in PD. However, transcranial ultrasonography does show hyperechogenicity in the lentiform nucleus in a significantly greater proportion of atypical parkinsonian syndromes than in PD.26 Transcranial ultrasound measurements demonstrate that the widths of the third ventricle and the frontal horn are smaller in multisystem atrophy (8.6 ± 1.5 mm and 15.1 ± 2.3 mm) than in PSP (12.9 ± 3.7 mm and 19.3 ± 4.2 mm), respectively.26
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


