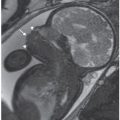
FIGURE 21.1: Cystic hygroma. Osteogenesis imperfecta type II at 14 weeks’ gestation. A: Sagittal US demonstrates subcutaneous edema along the cranium and spine. B: Coronal view demonstrates markedly short bowed humerus (cursers) and irregular beaded appearance of the ribs consistent with fractures.
1. Skull. Examination of the acoustic shadow behind the calvarium and the echogenicity of the bone itself allow evaluation for possible demineralization. Frontal bossing, craniosynostosis, cleft palate, micrognathia, or hypertelorism are features that may be seen in different SDs, and should be assessed when evaluating the fetus with a possible dysplasia.
2. Clavicle, scapula. A hypoplastic scapula may be seen in campomelic dysplasia (CD) or cleidocranial dysplasia; total or partial aplasia of the clavicles may occur in cleidocranial dysplasia.
3. Spine. The presence of segmentation anomalies, platyspondyly, abnormal curvature of the spine, and possible nonossification of the spine must be carefully evaluated.
4. Thorax. Detailed examination of the thorax is crucial, as chest restriction leads to pulmonary hypoplasia, a frequent cause of death in the neonate with SD (see below). A chest circumference measured at the level of the four-chamber heart of less than the 5th percentile for GA is a strong indicator of pulmonary hypoplasia. Other parameters indicative of pulmonary hypoplasia and/or lethality include chest circumference–abdominal circumference ratio less than the 5th percentile, chest–trunk length ratio less than 0.32, and a femur length–abdominal circumference ratio less than 0.16.9 Evaluation of the thorax should include a full analysis of the ribs, including number, shape, mineralization, and overall configuration.
5. Long bones. Evaluation of the long bones includes analysis of the degree of mineralization, presence of fractures or abnormal curvature of the long bones, as well as absence, hypoplasia, and malformation of the bone. Comparison among segments for shortening establishes whether the entity is a rhizomelic, mesomelic, or acromelic dysplasia, or if it involves all the segments of the extremities (micromelia) (Fig. 21.2).

FIGURE 21.2: Nomenclature for long bone shortening. A: Normal. B: Rhizomelia—shortening of the proximal segment. C: Mesomelia—shortening of the midsegments (radius and ulna/tibia and fibula). D: Micromelia—overall extremity length is small. Not depicted is acromelia, where the distal segments are short in length.
6. Pelvis. Abnormalities in the shape of the pelvis can provide clues as to the diagnosis of the SD at hand. A flared iliac wing may be found in entities such as Kniest dysplasia, a small pelvis may be seen in achondrogenesis or CD, and delayed ossification of the pubic bones may occur in ischiopatellar dysplasia.
7. Hands and feet. Polydactyly (more than 5 digits per hand) is classified as pre- or postaxial when the extra digits are located on the radial or ulnar side of the hand, respectively (Fig. 21.3). Other abnormalities, such as syndactyly (soft-tissue or bone fusion of adjacent digits), clinodactyly (deviation of the finger), postural deformities (“hitchhiker’s thumb,” “sandal toe deformity,” clinched hands), or abnormal extremities (broad tubular phalanges) must be evaluated (Fig. 21.4).
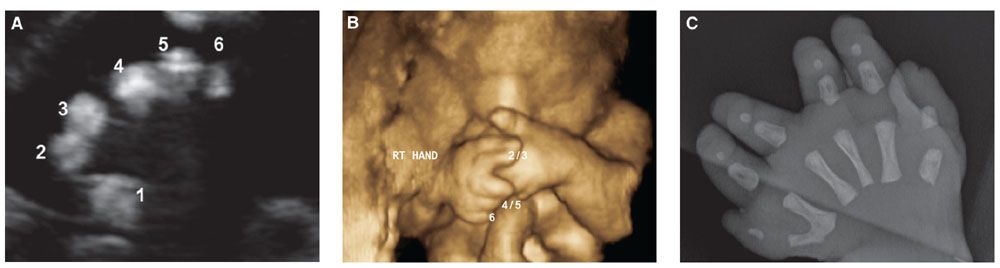
FIGURE 21.3: Polydactyly. Pallister–Hall syndrome at 28-week gestation. 2D (A) and 3D (B) US of the right hand demonstrate postaxial polydactyly and syndactyly of the 2nd and 3rd, as well as 4th and 5th digits. C: Postnatal radiograph of the hand confirmed the prenatal findings.
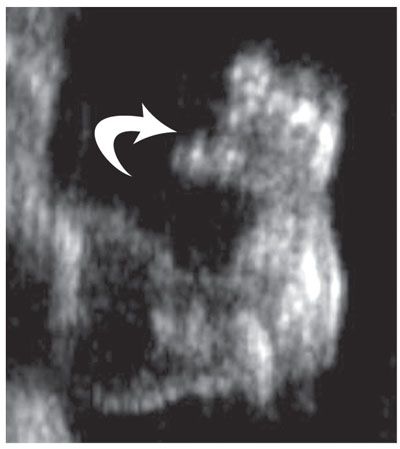
FIGURE 21.4: Diastrophic dysplasia. An 18-week fetus with short limbs. US of the hand demonstrates a classic “hitchhiker thumb” (arrow) with thumb abducted, helping confirm the diagnosis.
One of the most important determinations when evaluating the fetus with presumed dysplasia by US is the assessment and prediction of postnatal lethality. Lethality is mainly determined by the presence of pulmonary hypoplasia, whereby the small lungs are unable to maintain adequate oxygen exchange postnatally. Sonographic indicators of pulmonary hypoplasia are discussed.
Despite being the main imaging modality in the prenatal evaluation of the fetus, US has limited sensitivity in diagnosing SD prenatally. Diagnosis remains inaccurate with both false positive and false negative results. Published accuracy values range between 40% and 60%,10,11 opening the door to other imaging modalities to help confirm a diagnosis.
Magnetic Resonance Imaging
The use of magnetic resonance imaging (MRI) in the assessment of the fetal skeleton has advanced as technology improves.12,13 The development of fast echoplanar imaging (repetition time/echo time 3,000/45–60) has been particularly valuable in the evaluation of fetal cartilage and bone (Figs. 21.5 and 21.6). Connolly et al. documented the ability of MR to delineate cartilage transformation and development of secondary ossification centers in the fetal pig.14,15 Nemec et al.12 reviewed the long bone development of 253 fetuses from 19 to 36 weeks’ gestations by MR. In their series, the epiphysis changed in shape from spherical to hemispherical with a notch. The metaphysis was flat early in the second trimester and became undulated, while the perichondrium decreased in thickness with advancing gestational age. The range of diaphyseal lengths was wider than corresponding US values likely related to variability in fetal positioning.
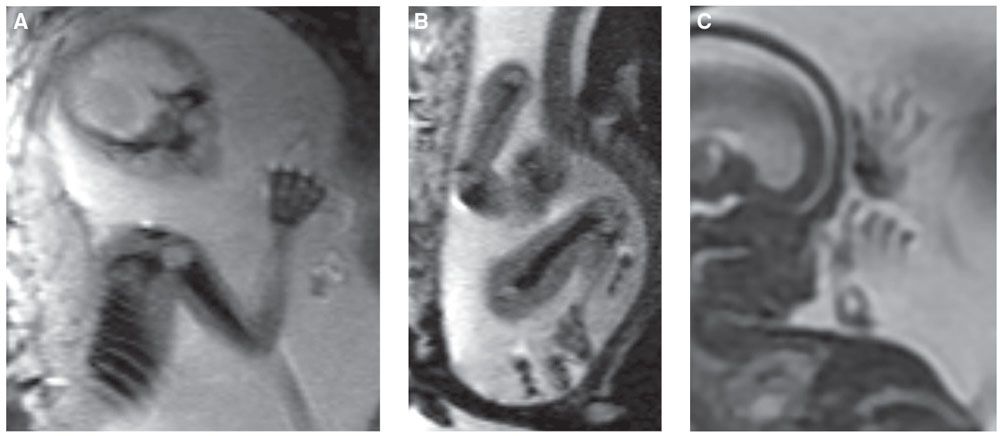
FIGURE 21.5: MRI of the fetal skeleton. Echoplanar image (EPI) of a 22-week fetus demonstrates low-signal skeleton and high-signal cartilage of the humerus, ribs, and hand (A) and humerus, femur, and portion of the tibia (B). C: SSFSE T2w MR image of a 17-week fetus demonstrates hands and fingers.
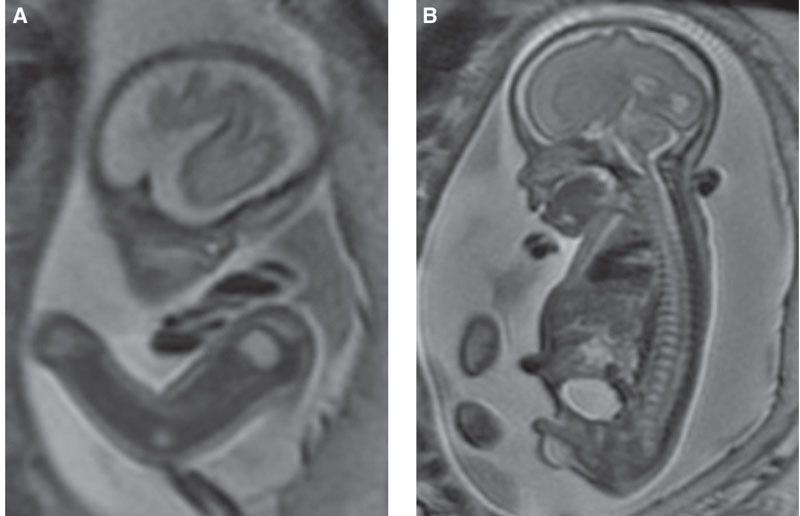
FIGURE 21.6: Kneist dysplasia. SSFSE T2w MR demonstrates unusually prominent high-signal epiphysis (A) and bright discs (B) in this fetus with moderate micromelia and platyspondyly. This nonlethal dysplasia is caused by mutations in COL2A1 with resultant joint contractures, scoliosis, and premature arthropathy.
Anecdotal cases of fetal MRI in the evaluation of specific SDs have been published.16–21 MR is particularly helpful when other anomalies are present such as cranial (craniosynostosis) or when oligohydramnios is present.20,21
Computed Tomography
The use of low-dose fetal CT in the evaluation of the fetal skeleton has developed as well.22–25 Given the obvious radiation dose to the fetus, this type of study is relegated exclusively to the fetus with severe osseous abnormalities, when the diagnosis is still in question and will impact outcome. Fetal CT should only be performed in the second or third trimester, when fetal organogenesis has already taken place.
When the decision is made to undergo this test, the patient is placed supine on the CT table. The top and bottom of the uterus are marked by US, providing the strict borders for CT scanning. The study is done without contrast and at such a low radiation dose that only the fetal bones (and not the solid organs) are visualized. Images are reviewed in the 3D console, following sectioning of the maternal structures, and rendering exquisite detail of the fetal skeleton (Figs. 21.7 to 21.9). By CT, the skull, clavicles, scapula, ribs, vertebral bodies, and long bones should be visualized in a normal fetus in the second and third trimester, and their absence implies pathology. Because of the low radiation used, the fetal hands and feet may not be seen, particularly in the second trimester; however, these are usually seen by US, obviating the need to increase the radiation dose to the fetus.
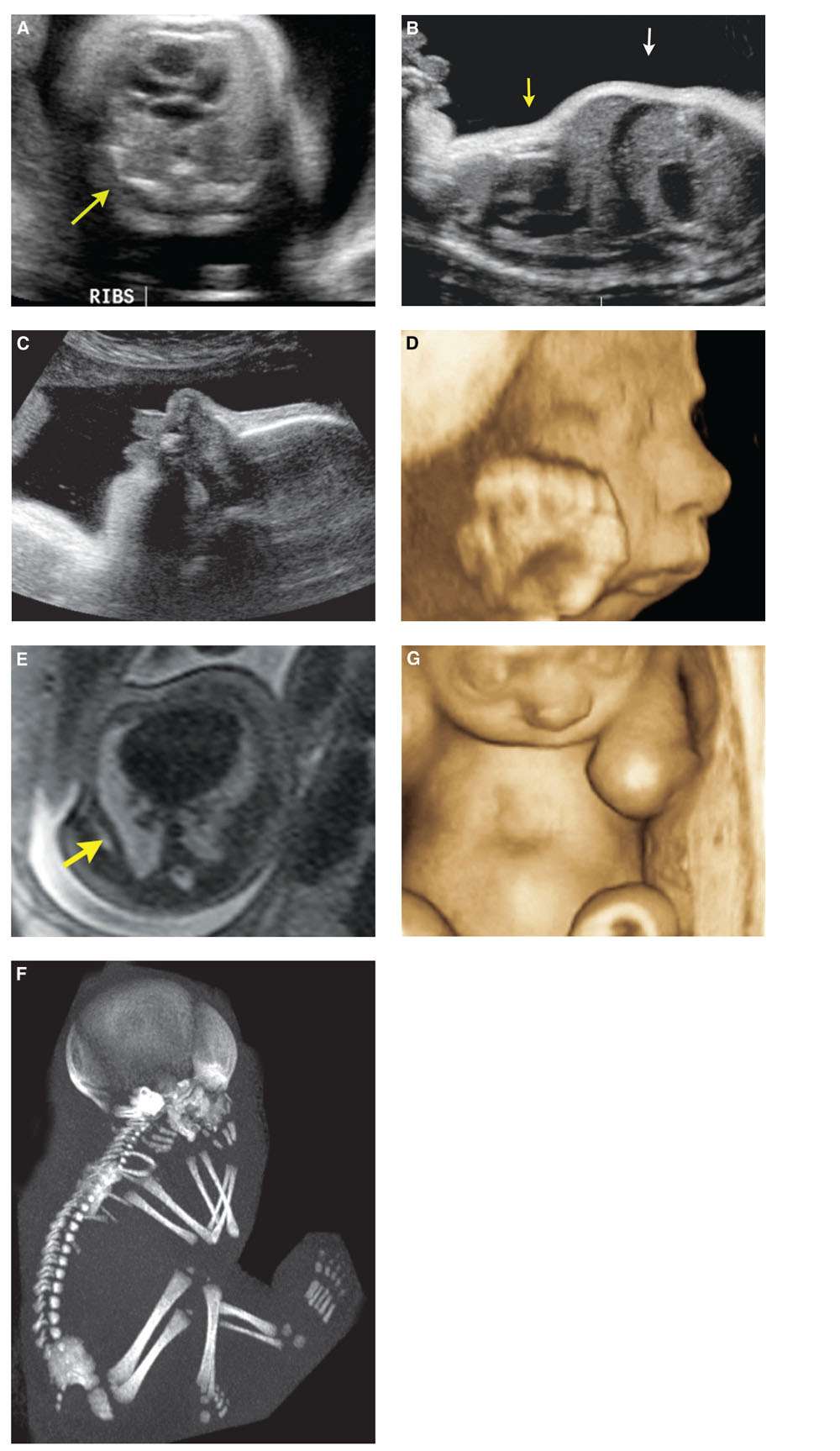
FIGURE 21.7: Cerebrocostomandibular syndrome. Thirty-four-week gestation with abnormally short ribs (arrow) (A), protuberant abdomen (white arrow), and small thorax (yellow arrow) (B). 2D (C) and 3D (D) US showed pronounced micrognathia. SSFSE T2 MR confirms short ribs (arrow) (E). Low-dose fetal CT demonstrates hypoplastic ribs with rib defects and gaps (F). 3D US reconstruction of the chest in the coronal plane depicts a pectus excavatum deformity (G).
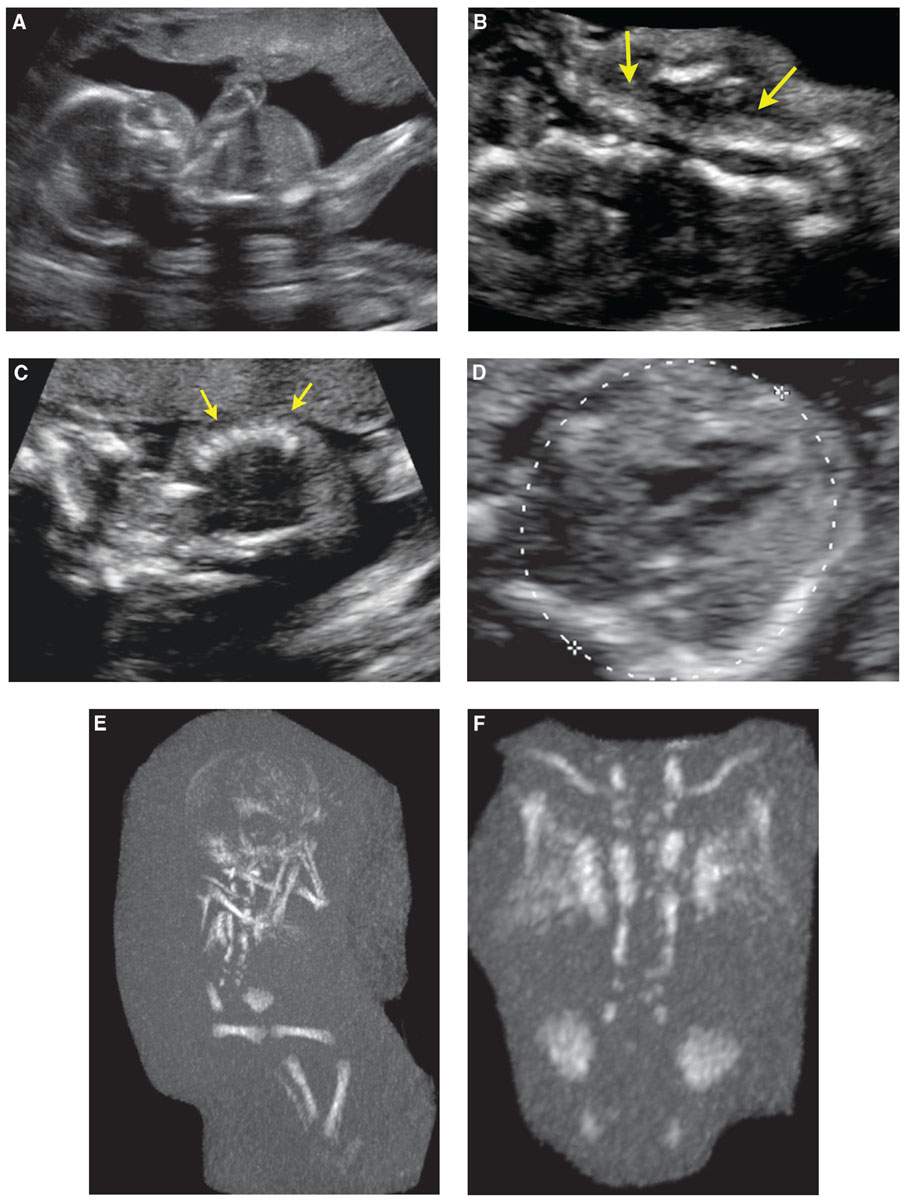
FIGURE 21.8: Spondylothoracic dysostosis. Twenty-one-week gestation with an overall short trunk and small chest/abdomen, on sagittal US (A). The vertebral bodies were disorganized (arrows in B). The ribs appeared close together (arrows) (C). In the axial plane, calipers are measuring the chest circumference, the ribs were short (D). the ribs were short (D). E: Low-dose fetal CT showed an overall short spine with normal-size long bones and head. The ribs were fused posteriorly and appeared to separate anteriorly, resembling a crab-like configuration. Magnified view of the thorax showed the ribs to better advantage (F). Also seen is a disorganized spine with multiple segmentation anomalies.
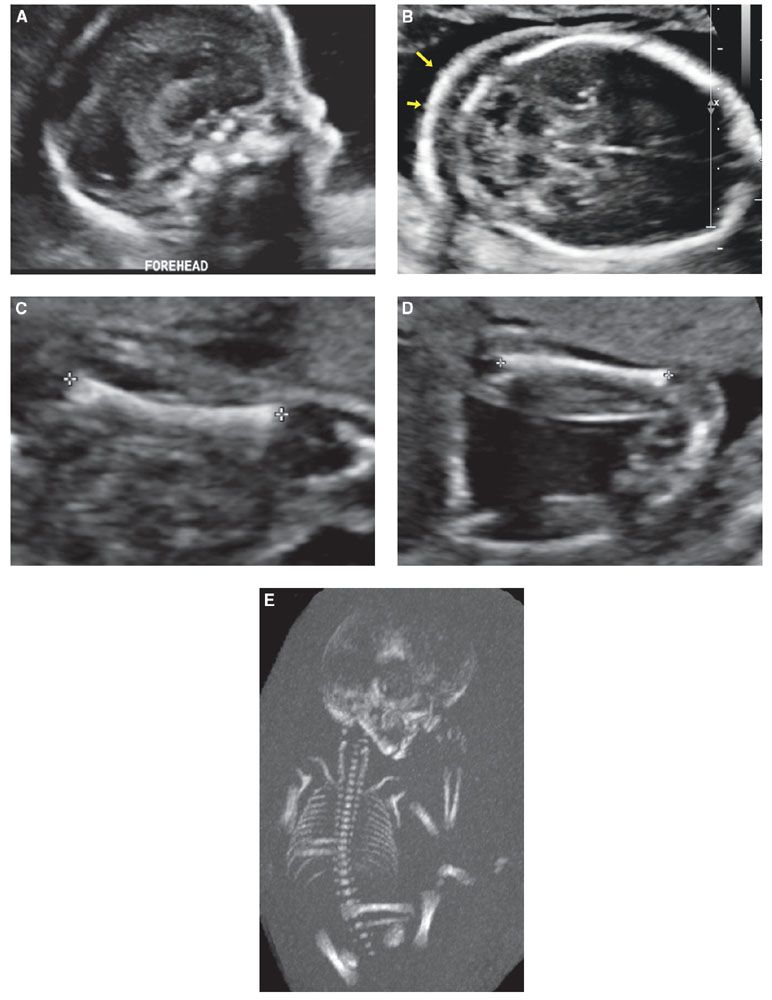
FIGURE 21.9: Pallister–Killian syndrome. Twenty-two-week fetus with non specific skeletal dysplasia, including: frontal bossing (A), nuchal thickening (arrows in B), short long bones without bowing or fractures (C), Flexion deformities at the wrist and ankles (D) were present by US. A normal-size rib cage was confirmed by low-dose fetal CT (E). Diagnosis of Pallister–Killian syndrome was confirmed molecularly with identification of isochromosome at 12p.
The analysis of a fetal CT should follow a systematic approach, as with US. It can demonstrate additional findings demonstrating bones in exquisite detail.25 Because of the radiation dose, however, fetal CT should be done only if the osseous findings are severe, and if the diagnosis in question is still uncertain by US.
There are some limitations in the evaluation of the fetus with SD when utilizing this imaging technique. First, the length of long bones in normal fetuses by CT has not yet been established. Secondly, assessment of mineralization currently is subjective. Using a fetal radiology atlas such as Schumacher et al.,26 which has postmortem radiographs of the normal fetus up to 23 weeks’ gestational age is useful as a reference. For older fetuses, mineralization can be assessed by comparing the skull with other cases of similar gestational age.
When performing a fetal CT, the goal is to use the lowest radiation dose possible while still having a diagnostically adequate study. We aim at a radiation dose of approximately 5 mSv. To put this in context, a study by McCollough et al.27 postulates that the absolute risk of fetal effects (including cancer induction) is small at a conceptus dose of 100 mSv, and negligible at a dose of less than 50 mSv. Assuming a normal incidence of 0.07% incidence of childhood cancer and a natural incidence of malformation risk in the order of 4% in the normal population, the risk to the conceptus given a fetal dose of about 50 mSv results in a 96% probability of birthing a child with no malformation, or a 99.9% chance that the child will not develop cancer, or 95.9% chance that the child will have no malformation nor cancer.22–28 The radiation risk to the fetus from a low-dose fetal CT is small, but real. Great care must be placed in proceeding with this type of study, and the decision should be taken by consensus by a group of radiologists, maternal fetal medicine specialists, and geneticists as well as the parents.
COMMONLY ENCOUNTERED PRENATAL SKELETAL DYSPLASIAS
Achondrogenesis
Differential Diagnosis: The term “achondrogenesis” (Greek for “not producing cartilage”) was coined by Marco Fraccaro, an Italian pathologist who observed a stillborn female with severe micromelia and marked histological changes of the cartilage.29 This is a lethal entity characterized by a relatively large head, short neck, extreme micromelia, and a defect in vertebral ossification. Achondrogenesis usually results in stillbirth or neonatal demise.
Incidence and Molecular Pathology: Birth prevalence is about 0.2 in 10,000 live births. It is inherited as autosomal recessive (type IA and IB) and autosomal dominant (type II). The locus for type IA is not yet known; the gene for type IB is responsible for a sulfate transporter, DTDST, located on chromosome 5q,29 also responsible for other forms of SDs including diastrophic dysplasia, atelogenesis type II, and multiple epiphyseal dysplasia. Type II achondrogenesis belongs to the type II collagenopathies and has a defect in the COL2A1 gene located in chromosome 12.
Radiologic Manifestations: Type I manifests with poorly mineralized skull, absent or minimal ossification of the vertebral bodies, pubis and sacrum, as well as short and thin ribs (with fractures, in the case of the fetus with type IA), short and deformed iliac wings, and marked shortness and bowing of the tubular bones. Type II has variable degrees of shortness of the long bones (from near normal to very short), lack of mineralization of many vertebral bodies, nonossified sacrum and ischium, small iliac wings with concave inferior and medial margins, and shortening of the ribs. Unlike type I, the calvaria demonstrates normal mineralization in type II achondrogenesis.
Diagnosis: Achondrogenesis can be diagnosed in the second trimester by US (Fig. 21.10). Limbs are abnormally short with small chest circumference. Unossified vertebrae may be missed sonographically if not carefully evaluated. At times, the fetus can present with hydrops (Fig. 21.11). Fetal CT can be helpful in confirming the unossified spine and short–long bones (see Fig. 21.10).
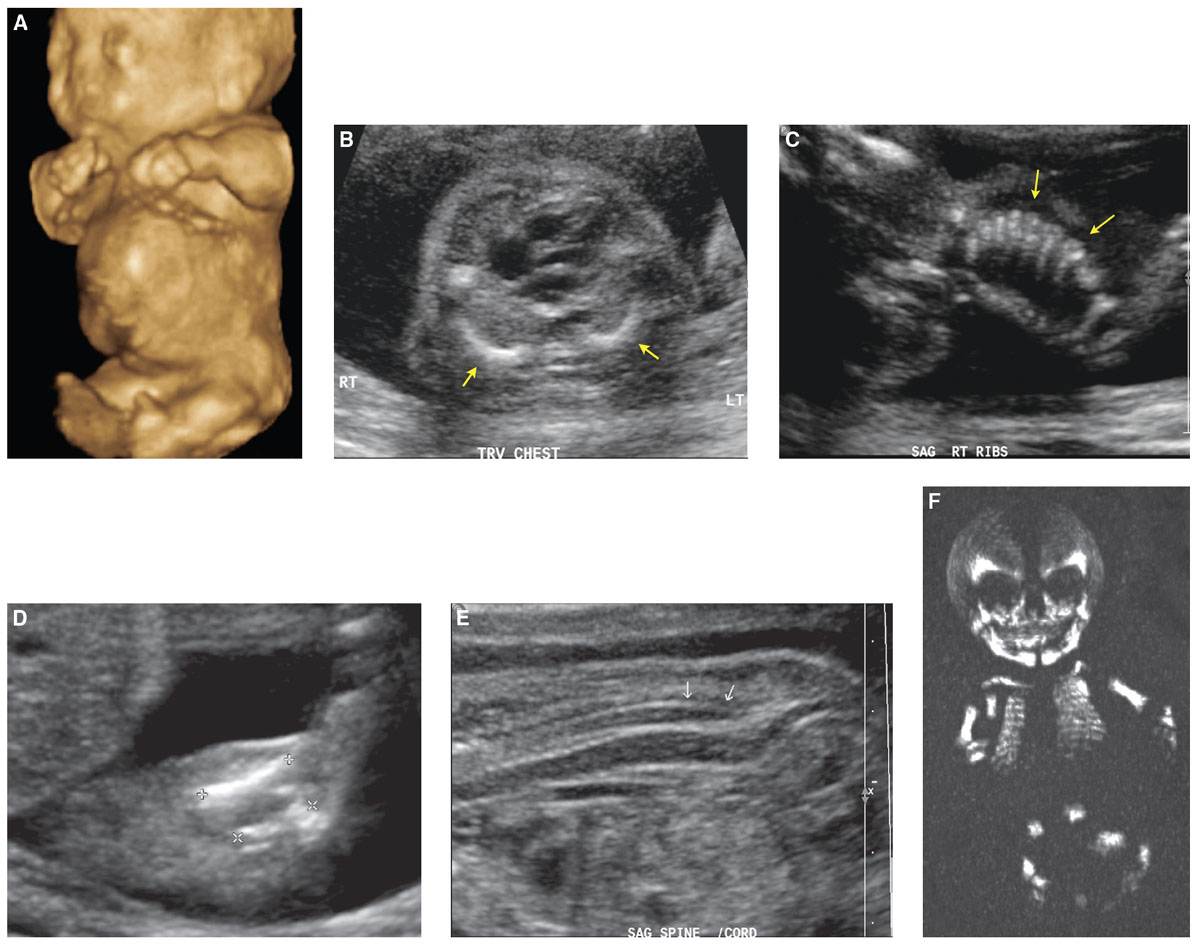
FIGURE 21.10: Achondrogenesis. 3D-US rendering of this 21-week fetus demonstrates shortening of the upper and lower extremities and a protuberant stomach (A). Axial sonographic image at the level of the heart shows markedly foreshortened ribs, denoted by arrows (B), which appear rather crowded and stacked (arrows) (C). The long bones are short (D). The spine is nonossified (arrows in E). Coronal 3D CT shows complete nonossification of the spine (F) with a “floating skull” appearance. The ribs and long bones are short. The iliac bones are small, and acetabula are flat.
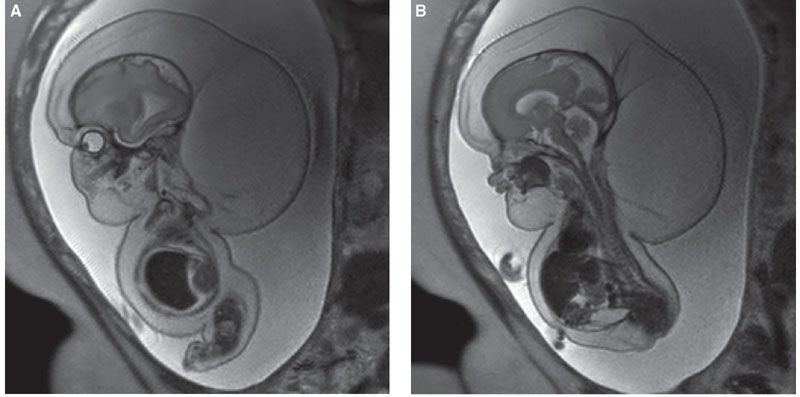
FIGURE 21.11: Achondrogenesis. A: SSFSE T2w MR demonstrates severe hydrops, hygroma, and ascites. The long bones are markedly short and bowed, the ribs short. B: The vertebrae are small and unossified.
Course and Prognosis: Achondrogenesis is uniformly lethal. If hydrops develops, in utero demise may occur. Polyhydramnios may be present and may accelerate delivery. Those that make it to term die in the neonatal period.
Differential Diagnosis: In achondrogenesis IB, there are no rib fractures, and the vertebral pedicles may be ossified, whereas type IA may have rib fractures. In achondrogenesis type II, the ribs are not as thin and show no fractures. Differential includes osteogenesis imperfecta (OI) and hypophosphatesia, which have absent or delayed ossification of the calvaria that is compressible.
Achondroplasia
Description: The term “achondroplasia” was used in the past to define all short-limb dysplasias. With recognition of the heterogeneity of these disorders, this term is currently used to describe a specific disease characterized by predominantly rhizomelic dwarfism, limb bowing, lordotic spine, bulky head, and depressed nasal bridge.
Incidence and Molecular Pathology: It is the most common nonlethal SD (approximately 1 in 25,000 births) that may be transmitted as an autosomal dominant pattern or, in 80% to 90% of cases, a spontaneous mutation. It is caused by a gain-of-function mutation in the fibroblast growth receptor 3 gene (FGFR3), a transmembrane tyrosine kinase receptor that binds fibroblast growth factors. The gene is located in the short arm of chromosome 4 and codifies a highly conserved protein. The most common mutation, a G>A transition at nucleotide 1,138 (Gly380Arg), is observed in 95% of affected patients. The gain-of-function mutation causes ligand-independent activation of FDFR3, resulting in inappropriate cartilage growth plate differentiation and deficient endochondral growth and ossification.30 Targeted molecular confirmation can be carried out using cell-free fetal DNA.31
The homozygous form is more severe, resembling thanatophoric dysplasia and is lethal.
Radiologic Manifestations: Achondroplasia is a rhizomelic micromelia with mild bowing of the long bones and disproportionately large head. The nasal bridge is depressed, there is frontal bossing and decreased size of the skull base and foramen magnum. Hydrocephalus may develop. Short, flat vertebral bodies that may be bullet-shaped, progressive narrowing of the interpediculate distance in the lumbar spine, square iliac wings, short narrow sciatic notch, and flat acetabular roofs are findings encountered in achondroplasia. A trident hand with fingers of equal length and separation of the third and fourth fingers may be present.
Diagnosis: Heterozygous achondroplasia is typically not diagnosed in the first and early second trimester. Femoral length crosses below the 3rd percental only after 18 to 26 weeks gestation with the affected fetus having relatively normal long bone growth before then.32 Establishment of a femoral growth curve in the second trimester helps to distinguish between homozygous, heterozygous, and unaffected fetuses. Fetal size charts show femoral length on or below the 3rd percentile by 25 weeks’ gestation and always below the 3rd percentile by 30 weeks.31 The head circumference increases to above the 95th percentile in a majority of cases, with a slight increase in abdominal circumference as well. Additional US findings include frontal bossing, slightly bowed femora, short fingers, small chest, and polyhydramnios (Fig. 21.12).
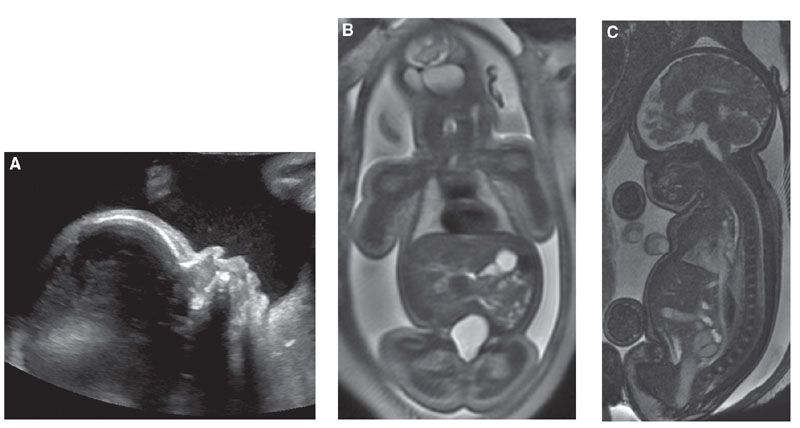
FIGURE 21.12: Achondroplasia. A: Sagittal US demonstrates frontal bossing at 32 weeks’ gestation. B: Coronal SSFSE T2w MR demonstrates short humeri but normal lung volume. C: Sagittal SSFP MR confirms frontal bossing and documents narrowing of the foramen magnum.
Fetal MRI can help confirm the diagnosis documenting narrowing of the foramen magnum (Fig. 21.12C).
Homozygous achondroplasia can be diagnosed much earlier due to the presence of severe micromelia. Femoral length will fall below the 3rd percentile by 14 to 16.5 weeks’ gestation.33,34
Course and Prognosis: Intelligence and life span are usually normal. Postnatal complications are mainly neurologic and are related to the degree of foramen magnum narrowing and spinal cord compression.
Homozygous achondroplasia (when both parents are affected) is lethal, with either stillbirth or early neonatal death from respiratory failure.
Differential Diagnosis: Differential diagnosis includes asymmetric IUGR and familial short stature. Assessment for fetal health and family history is important in distinguishing between these entities. Hypochondroplasia is a relatively common milder form that varies within and between families and lacks the cranial complications with lack of frontal bossing.35
Fetuses with the severe form of homozygous achondroplasia present early in the second trimester with severe micromelia. In these cases, other lethal SDs such as thanatophoric should be included in the differential and correlated with family history.33,34
Arthrogryposis
Description: Arthrogryposis multiplex congenital (AMC) is a descriptive term that refers to multiple joint contractures present at birth. This term is often misused as a specific diagnosis instead of a descriptor or clinical finding. Arthrogryposis is not a specific disorder nor a SD, but the consequence of neurological, muscular, connective tissue, and skeletal abnormalities or intrauterine crowding, which may lead to limitation of fetal joint mobility and subsequent development of contractures. Potential etiologies include neuropathic abnormalities (brain, spine, or peripheral nerve), abnormalities of muscle structure or function (muscular dystrophies, mitochondrial abnormalities), abnormalities of connective tissue (diastrophic dysplasia), space limitation (multiple gestations, oligohydramnios), maternal diseases (multiple sclerosis, myasthenia gravis), and impaired intrauterine or fetal vascularity.36 The common outcome is the decrease or lack of fetal movement leading to collagen proliferation, fibrotic replacement of the muscle, and marked thickening of the joint capsules.
Incidence and Molecular Pathology: Approximately 1% of all live births demonstrate some type of contracture involving one or more joints. The overall incidence of arthrogryposis, with multiple joint involvements, is 1 in 3,000 live births.37 Arthrogryposis may be caused by genetic and environmental factors. Among the genetic factors, more than 35 specific genes have been associated with this condition. Since the evaluation of every single one is beyond the scope of this article, the reader is directed to a comprehensive review of the genetic etiologies.38
Diagnosis
Ultrasound: Decreased fetal motion as detected by US during the second or third trimester or by maternal perception may be the first clue in detecting prenatal arthrogryposis. Real-time sonographic evaluation may show contractures with decreased fetal motion or severe flexion/extension deformities, unchanged in appearance during the time of scanning (Fig. 21.13).
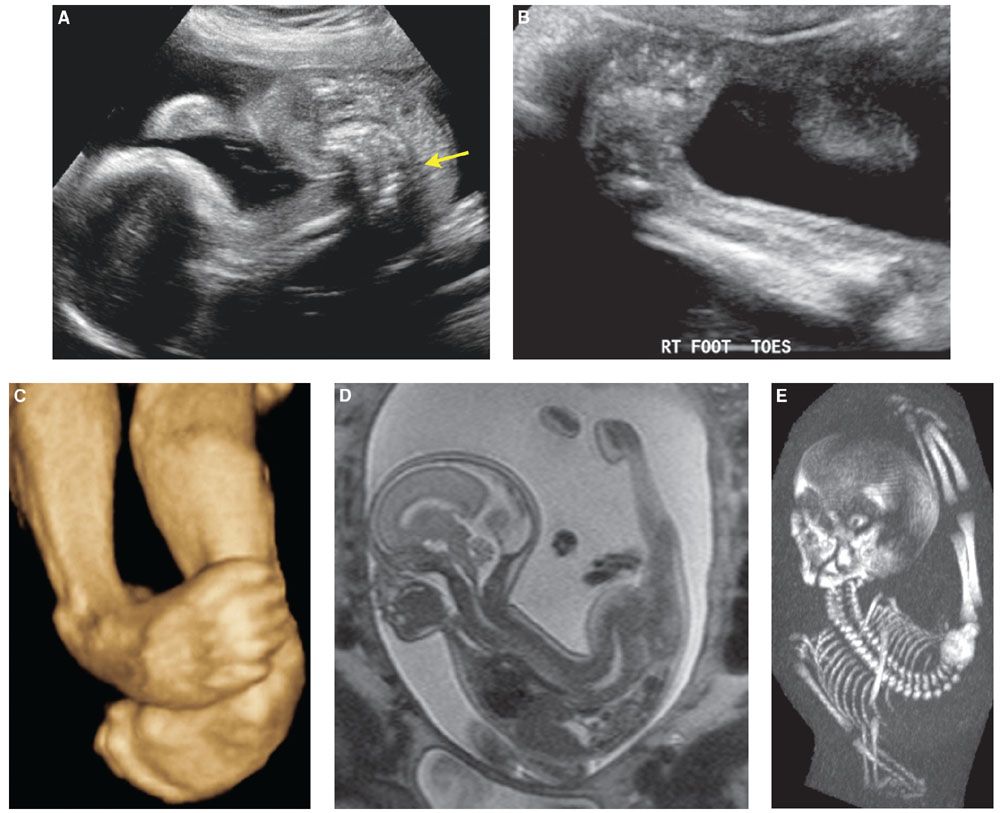
FIGURE 21.13: Arthrogryposis. 2D sonographic view of this 23-week fetus demonstrates a severe lumbosacral lordosis (arrow in A). Bilateral clubfeet are shown in this 2D image (B) and 3D US rendering (C). SSFSE T2w sagittal MR image of the fetus demonstrates the scoliosis with marked extension contractures of the lower extremities and partial visualization of the clubfoot deformity (D). Low-dose fetal CT shows abnormal spinal curvature and the abnormal posturing of the extremities (E).
MRI: MR cine sequences may be helpful in evaluating fetal movement as well. Fetal MR can help delineate intracranial and/or spine pathology that may be directly responsible for decreased fetal movement. Subjective evaluation of muscle mass can be provided by both US and MR.
CT: Fetal CT may provide 3D rendering of the bones that may be more easy to visually relate to for parents, obstetricians, and orthopedic surgeons (Fig. 21.13E).
Course and Prognosis: The prognosis is related to the specific etiology. In some severe cases, the underlying disease is lethal, with death occurring shortly after birth. In other cases, musculoskeletal impairment is minimal, and intelligence is normal. In between these two extremes, infants may have different degrees of disabilities. It is estimated that up to 50% of children born with arthrogryposis and CNS involvement will die in the neonatal period.36 For those with contractures, treatment is directed toward achieving stable functional weight-bearing and manual dexterity.
Asphyxiating Thoracic Dysplasia
Synonym: Jeune syndrome
Description: This autosomal recessive osteochondrodysplasia is grouped among the short-rib dysplasias including the lethal short-rib polydactyly syndromes (SRPS I–V) and less severe Ellis–van Creveld syndrome. These short-rib dysplasias (with or without polydactyly) appear to share a common primary cilium function defect and form a subset of ciliopathy disease. Jeune and Ellis–van Creveld have overlapping but milder phenotype as compared with the SRPS 1–V.39
Characteristic skeletal abnormalities include small, narrow thorax, short ribs, short squared iliac wings, and limb shortening. The spectrum of clinical manifestations is wide, ranging from lethal pulmonary hypoplasia to mild forms without respiratory compromise. Renal, hepatic, pancreatic, and retinal complications may also occur. Renal hypertension and failure have been reported findings. Hepatic involvement may include prolonged neonatal jaundice, polycystic liver disease, and hepatic cirrhosis. Pancreatic cystic disease may be present. Retinal abnormalities include retinal dystrophy and abnormalities in pigmentation. Other pathologic findings, including agenesis of the corpus callosum or Dandy Walker malformation, may be present.
Incidence and Molecular Pathology: The estimated prevalence is 0.14 per 10,000 births. Most mild and severe forms map to chromosome 15q13.40 The causative gene has not yet been found.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


