IDIOPATHIC INTERSTITIAL PNEUMONIAS
IDIOPATHIC INTERSTITIAL PNEUMONIAS
The term idiopathic interstitial pneumonia (IIP) is applied to a group of inflammatory and fibrotic lung diseases with distinct histologic and imaging appearances, and without a known cause. The concept of interstitial pneumonia originated with Liebow and Carrington, and the first comprehensive and critical attempt to subdivide IIP based on histologic changes was made by Liebow, 1 dividing these cases into five groups (Box 10.1). Over time, this list has been repeatedly revised.2.3. and 4. Giant cell interstitial pneumonia (GIP) is no longer included as it is usually a manifestation of hard metal pneumoconiosis.5. and 6. Bronchiolitis interstitial pneumonia was renamed as cryptogenic organizing pneumonia (COP); 7 the alternative term bronchiolitis obliterans organizing pneumonia (BOOP) 8 is no longer recommended. 4 Although most cases of lymphoid interstitial pneumonia (LIP) are secondary to other underlying conditions such as acquired immune deficiency syndrome (AIDS) or Sjögren syndrome (SjS),9. and 10. LIP is still included in the classification. The newly recognized entities of acute interstitial pneumonia (AIP) and nonspecific interstitial pneumonia (NSIP) have been added.
Box 10.1
• Usual interstitial pneumonia (UIP)
• Desquamative interstitial pneumonia (DIP)
• Bronchiolitis obliterans with interstitial pneumonia (BIP)
• Lymphoid interstitial pneumonia (LIP)
• Giant cell interstitial pneumonia (GIP)
None of the previous classifications of the IIPs1.2. and 3. has clearly delineated the complementary roles of the pathologist, radiologist, and clinician in diagnosing these conditions. Because of this, and because of substantial variation in definition and terminology of the IIPs, the American Thoracic Society (ATS) and the European Respiratory Society (ERS) convened an international committee of pulmonologists, thoracic radiologists, and pulmonary pathologists to clarify the nomenclature and typical patterns of these conditions.4. and 11. The interstitial pneumonias included in this classification are usual interstitial pneumonia (UIP), NSIP, AIP, desquamative interstitial pneumonia (DIP) and its related entity respiratory bronchiolitis interstitial lung disease (RB-ILD), LIP, and COP (Table 10.1; Box 10.2). The key concepts underlying the classification are as follows (Box 10.3):
• The classification is based on histologic criteria, but there is a clear recognition that the CT pattern is important in delineating the macroscopic morphology of the IIPs.
• Each pattern of interstitial pneumonia may be idiopathic or may be secondary to an identifiable cause such as collagen vascular disease, drugs, or inhalation exposure. Careful clinical evaluation is required to identify an underlying cause for the interstitial pneumonia. The classification clearly separates the morphologic pattern identified by the pathologist and radiologist (which may or may not be idiopathic), from the idiopathic clinical syndrome identified by the clinician. For example, the term idiopathic pulmonary fibrosis (IPF) is reserved for the idiopathic clinical syndrome associated with the morphologic pattern of UIP. The pathologist and radiologist are encouraged to use the term ‘pattern’ when referring to morphologic findings, to emphasize the fact that these morphologic patterns may be due to a variety of types of lung injury.
• The clinician, radiologist, and pathologist have complementary roles in diagnosis of interstitial pneumonias. The final diagnosis is made by integration of the clinical, imaging, and pathologic features.
| Morphologic entity | Associated clinical syndrome | Histologic findings | CT features and distribution | Radiologic differential diagnosis |
|---|---|---|---|---|
| Usual interstitial pneumonia (UIP) | Idiopathic pulmonary fibrosis (IPF) | Heterogeneous areas of young connective tissue, scarring, honeycombing, and normal lung; fibroblastic foci; patchy, often subpleural | Reticular abnormality; honeycombing; often patchy; basal, peripheral predominance | Collagen vascular disease; asbestosis; chronic hypersensitivity pneumonitis; NSIP |
| Nonspecific interstitial pneumonia/fibrosis (NSIP) | Nonspecific interstitial pneumonia | Alveolar septal thickening by inflammation or fibrosis; spatially and temporally homogeneous | Ground-glass abnormality; reticular abnormality; traction bronchiectasis; basal predominance ± subpleural sparing | Collagen vascular disease; asbestosis; chronic hypersensitivity pneumonitis; DIP, OP |
| Organizing pneumonia (OP) | Cryptogenic organizing pneumonia (COP) | Intraluminal organizing fibrosis in bronchioles, alveolar ducts, and alveoli; temporally homogeneous; patchy distribution | Consolidation, ground-glass abnormality; patchy; basal, peripheral/ peribronchovascular predominance | Infection; vasculitis; sarcoidosis; lymphoma; bronchioloalveolar cell carcinoma; NSIP cell |
| Desquamative interstitial pneumonia (DIP) | Desquamative interstitial pneumonia (DIP) | Diffuse macrophage accumulation within alveolar spaces; mild interstitial thickening; homogeneous involvement | Ground-glass attenuation; ± cysts; basal, peripheral predominance | Hypersensitivity pneumonitis; NSIP; LIP |
| Respiratory bronchiolitis interstitial lung disease (RB-ILD) | Respiratory bronchiolitis interstitial lung disease (RB-ILD) | Bronchiolocentric accumulation of alveolar macrophages; mild bronchiolar fibrosis | Centrilobular nodules; ground-glass attenuation; diffuse or upper lung predominance | Hypersensitivity pneumonitis |
| Acute interstitial pneumonia (AIP) | Acute interstitial pneumonia (AIP) | Acute: edema, hyaline membranes; interstitial inflammation; organizing: airspace organization | Acute: ground-glass, consolidation; organizing: architectural distortion, traction bronchiectasis | Hydrostatic edema; pneumonia; acute eosinophilic pneumonia |
| Lymphoid interstitial pneumonia (LIP) | Lymphoid interstitial pneumonia (LIP) | Diffuse alveolar infiltration by lymphocytes; frequent lymphoid hyperplasia | Centrilobular nodules, ground-glass attenuation, septal thickening; cysts; diffuse or lower lung distribution | Sarcoidosis; Langerhans cell histiocytosis; NSIP; DIP |
Box 10.2
• UIP: usual interstitial pneumonia
• NSIP: nonspecific interstitial pneumonia
• DIP: desquamative interstitial pneumonia
• RB: respiratory bronchiolitis
• RB-ILD: respiratory bronchiolitis interstitial lung disease
• OP: organizing pneumonia
• COP: cryptogenic organizing pneumonia
• AIP: acute interstitial pneumonia
• LIP: lymphoid interstitial pneumonia
• IPF: idiopathic pulmonary fibrosis
Box 10.3
• Patterns of IIP are defined on the basis of their histologic features
• Each IIP has an associated prototypic CT appearance
• Each pattern of pneumonia can be associated with other causes, particularly collagen vascular disease
• Multidisciplinary evaluation by clinician, radiologist, and pathologist is important.
Histologically, UIP is the most common of the idiopathic interstitial pneumonias.12.13.14. and 15. NSIP and COP are relatively common, while AIP, DIP, RB-ILD, and LIP are relatively rare.
The 5-year survival in patients with histologic UIP ranges from 15% to 40%, compared with 60% to 100% for NSIP, and 100% for DIP.12.14.15. and 16. Because of these substantial differences in survival, it is important to clearly separate those patients who have the UIP pattern on histology or imaging from those with other patterns. In those patients who do not undergo biopsy, the CT pattern becomes central in making this distinction.
The emergence of the current classification has depended critically on examination of sizeable specimens of lung (obtained by thoracoscopy or thoracotomy), allowing assessment not only of the nature of the inflammation and fibrosis but also the spatial and temporal distribution of lesions. 3 Nevertheless, it needs to be recognized that a biopsy specimen, however generous, may not be wholly representative and that different histopathologic subtypes can coexist in a given patient. 17 For these reasons, evaluation by CT is important to determine whether the dominant imaging pattern is consistent with the histologic diagnosis.
Before the publication of the ATS statement on IPF, 18 and the ATS/ERS classification of idiopathic interstitial pneumonias, 4 the terms ‘idiopathic pulmonary fibrosis’ (in the USA),19. and 20. ‘cryptogenic fibrosing alveolitis’ (in the UK or Canada),21.22.23. and 24. or ‘idiopathic interstitial pneumonia’ (in Asia) 25 were generally used to refer to patients who presented with an idiopathic clinical syndrome of progressive shortness of breath associated with physiologic and imaging evidence of ILD. On review of the biopsies of such patients using current criteria, the histologic diagnosis is UIP in 50–60% of cases, NSIP in 15–35%, and DIP or RB-ILD in 10–15%.12.13.14.15. and 16. Case series of such patients published prior to 2000 may include unknown proportions of UIP, NSIP, DIP, and RB-ILD. For this reason, descriptions of the imaging appearances of these patients published before 2000 are difficult to interpret.
Thoracoscopic lung biopsy is not usually indicated in individuals with typical clinical and computed tomography (CT) features of UIP. 4 However, biopsy is recommended in all patients without such a typical appearance, because the distinction between UIP, NSIP, and the other entities is of substantial prognostic importance. Despite this recommendation, many patients with CT appearances of lung fibrosis do not undergo biopsy, for reasons ranging from medical frailty to lack of availability of trained pulmonary pathologists. It is hoped that increasing awareness of the influence of histology on prognosis, and wide availability of video-assisted thoracoscopy for biopsy, should increase the biopsy rate in patients without a definitive diagnosis on CT. In patients who do not undergo biopsy, high-resolution CT (HRCT) assumes a central role in defining the morphologic pattern, and may serve as a surrogate for biopsy. 16
Clinical presentation
The most frequent presenting symptoms of IIP are progressive exertional dyspnea and cough, which is usually nonproductive but in one series was productive in just over half of the patients. 26 Less common symptoms include nonspecific chest pain, 27 and constitutional symptoms such as fever, weight loss, and fatigue. 28 A nonerosive arthropathy that is not part of a recognized connective tissue disorder is quite common.
Changes in pulmonary function tests26.28. and 29. are those that might be expected from a diffuse interstitial process: reduced compliance and lung volumes, particularly vital capacity and total lung capacity with relatively normal residual volume. Reduction in gas diffusing capacity is a particularly early and characteristic change. In patients with coexisting emphysema, lung volumes may appear preserved, and in such individuals lung gas diffusing capacity is usually precipitously decreased.30.31. and 32.
As discussed below, all of the patterns of interstitial pneumonia are strongly associated with collagen vascular disease. Features suggestive of collagen vascular disease, such as an isolated arthropathy, Raynaud syndrome, or autoantibodies, are seen in many patients with interstitial pneumonia who do not meet criteria for collagen vascular disease. Some of these patients will later develop a full-blown collagen vascular disease. Overall more than half the patients with the syndrome of IIP will have autoantibodies, with antinuclear antibody positive in 15–45% of patients, and rheumatoid factor present in up to a third.26.33. and 34. Available evidence suggests that patients with interstitial pneumonia related to collagen vascular disease have a better prognosis than those with IIP, 35 but much if not all of this difference is likely due to the fact that NSIP is the predominant histology in patients with collagen vascular disease, particularly in scleroderma.36. and 37.
Cigarette smoking confers an increased risk for the subsequent development of lung fibrosis, with an odds ratio of about 1.6 compared with nonsmokers, increasing with heavier smoking. 38 Epidemiological studies have suggested that prolonged metal or wood dust exposure might account for up to 20% of cases of lung fibrosis, 39 and that certain prescribed drugs (particularly antidepressants) may be responsible for approximately 11% of cases. 40
The clinical and imaging evaluation of the patient with a syndrome of interstitial pneumonia should focus on two questions:
• Is it idiopathic? Interstitial pneumonia related to a recognizable cause such as hypersensitivity pneumonitis, asbestos exposure, drugs, or collagen vascular disease may be managed differently and may have a different prognosis from IIP. While clinical and laboratory evaluation is important in making this distinction, CT can yield important clues, particularly in raising the suspicion of hypersensitivity pneumonitis or collagen vascular disease (see Figs 10.27 and 10.29 below).
• Does the patient have a morphologic pattern of UIP? The morphologic pattern of UIP is usually refractory to standard anti-inflammatory treatments, and is associated with a significantly worse prognosis than the other interstitial pneumonias. Imaging is often pivotal in making this distinction (Table 10.2).
| CT features suggesting the diagnosis of UIP | Features suggesting that the diagnosis is probably not UIP |
|---|---|
| Lower lung predominance, though upper lungs are usually also involved | Mid- or upper lung predominance |
| Peripheral predominance | Peribronchovascular predominance |
| Honeycombing | Extensive ground-glass abnormality |
| Centrilobular nodules | |
| Marked septal thickening | |
| Non-honeycomb cysts | |
| Multiple lobules of mosaic attenuation or expiratory air-trapping |
Usual interstitial pneumonia/idiopathic pulmonary fibrosis
Histopathology
UIP is the most common histopathologic pattern seen in patients who present with the clinical syndrome of IIP.3.12.13.14.15. and 41. Under the ATS/ERS classification, the term idiopathic pulmonary fibrosis is applied exclusively to the idiopathic clinical syndrome associated with the morphologic pattern of UIP.4. and 18. The key histopathologic features of UIP (Box 10.4) are: 42
• Patchy and variable distribution of areas of fibrosis at different stages of maturity, often with islands of normal lung interspersed with advanced fibrosis
• Presence of architectural distortion with honeycombing or scars
• Presence of small areas of young connective tissue, called fibroblastic foci.
Box 10.4
• Fibroblastic foci
• Dense fibrosis
• Honeycombing
• Paucity of inflammatory cells
• Temporal and spatial nonuniformity
The variation in types of lung fibrosis is most obvious at low magnification, with alternating areas, from one field to another, of young connective tissue, interstitial fibrosis, honeycomb destruction, and normal lung. Within areas of honeycomb lung, the enlarged airspaces are lined by bronchiolar epithelium and hyperplastic alveolar pneumocytes; within the airspaces there may be mucus and inflammatory cells (most frequently neutrophils). Other pathologic features encountered in UIP are endarteritis obliterans and smooth muscle hypertrophy, but these are not specific to UIP and are probably secondary phenomena.
The areas of young connective tissue seen on biopsy in UIP consist of small foci of actively proliferating fibroblasts and myofibroblasts.43. and 44. These fibroblastic foci, representing ongoing sites of lung injury and repair, are thought to represent the earliest lesion of UIP, and are very important for the histologic diagnosis of UIP. 4 They may represent areas of lung response to an injury that is as yet undefined.45. and 46. They often occur at the interface between fibrotic and normal lung. The extent of fibroblastic foci on lung biopsy is an important predictor of survival in patients with UIP. 47
The recognition of fibroblastic foci as the important pathogenic lesions of UIP represents an important paradigm shift from the previous concept of alveolar inflammation or alveolitis as the precursor of UIP. 41 A chronic inflammatory cell infiltrate may indeed be seen in UIP, but is usually mild and consists mainly of lymphocytes with occasional plasma cells and eosinophils. If extensive cellularity is found, the diagnosis is probably not UIP. The extent of alveolar cellularity does not predict survival. 47 The paucity of inflammatory infiltration of the lung, in contrast to the other idiopathic pneumonias, may explain the lack of steroid responsiveness in UIP/IPF.
Causes of a UIP pattern on histology and CT (Box 10.5) include chronic hypersensitivity pneumonitis, collagen vascular disease, familial pulmonary fibrosis, and asbestos exposure.4. and 48. Drug toxicity is a rare cause of a UIP pattern, best described with nitrofurantoin. 49
Box 10.5
• Idiopathic pulmonary fibrosis
• Collagen vascular disease
• Hypersensitivity pneumonitis
• Asbestosis
• Familial
• Drugs (very rare)
The pathogenesis of UIP remains unclear. The fibroblastic foci are thought to represent an abnormal wound healing response to lung injury, resulting from release of multiple fibrogenic cytokines. 41 The inciting agent remains unclear. There has been much speculation about the possible role of viruses, including the Epstein–Barr virus, hepatitis C virus, herpesvirus, and adenoviruses, in the initiation of IPF, but no specific relationship has been proved.50.51.52.53. and 54.
Epidemiology and clinical presentation
UIP is more common in men than in women (1.5–1.7 : 1).21.38. and 55. Most patients are over 50 at the time of diagnosis, and about two-thirds are over 60, 55 but the disease may occur in people as young as 28.3. and 56. There have been descriptions of UIP occurring in children and adolescents, but these entities would probably not meet the current criteria for diagnosis of UIP, 3 and no cases of UIP were recorded in two series of 51 and 38 pediatric patients with diffuse lung disease that used current diagnostic criteria.57. and 58.
Patients with UIP usually complain of a dry cough and progressive exertional dyspnea of insidious onset. Systemic symptoms such as fever, weight loss, and arthralgia occur with variable frequency, 29 although fever itself is unusual and probably occurs in less than 15% of patients.43. and 59. Some patients date the onset of symptoms to an influenzalike illness. 28 A small proportion of patients present without symptoms, because of an abnormal radiograph or CT scan. 60 On physical examination, late, fine inspiratory crackles at the lung bases are an almost universal finding, 26 and two-thirds to three-fourths of the patients show clubbing of the fingers.26.27.60. and 61. Occasionally, there is full-blown hypertrophic osteoarthropathy. 27 In advanced disease cor pulmonale and cyanosis may develop. 28
The prognosis of patients with biopsy-proven UIP is poor, with mean survival after presentation ranging from 2.8 years to 9 years.12.26.62. and 63. At 5 years after diagnosis, only 12%15 to 45% of patients with UIP survive.12.14. and 64. Lower mortality rates were reported in earlier series,65.66. and 67. but it is likely that these included other IIP subtypes such as NSIP. The survival of patients with UIP is clearly worse in recent series which have excluded patients with NSIP and DIP.14.15.16. and 68.
Respiratory failure is the cause of death in 50–90% of patients with IPF.69.70. and 71. The majority of the respiratory deaths are related to acute exacerbation of UIP.69.70. and 72. The second most common cause of death is cardiovascular disease, while lung cancer accounts for about 10% of deaths.71. and 72. Given the high mortality, there is substantial interest in developing a treatment for IPF. There is no evidence that immunosuppressive therapy or corticosteroids are effective in UIP, although these agents are still commonly used. 73N-Acetylcysteine may help prevent decline in pulmonary function. 74 Bosentan, an endothelin antagonist, showed a trend toward delayed time to death or disease progression: 75 this effect was more pronounced in those who had undergone biopsy, and particularly in patients who did not have honeycombing on CT at presentation. 76 Other potentially helpful agents currently undergoing evaluation include sildenafil77 and perfenidone. 78
Imaging appearances
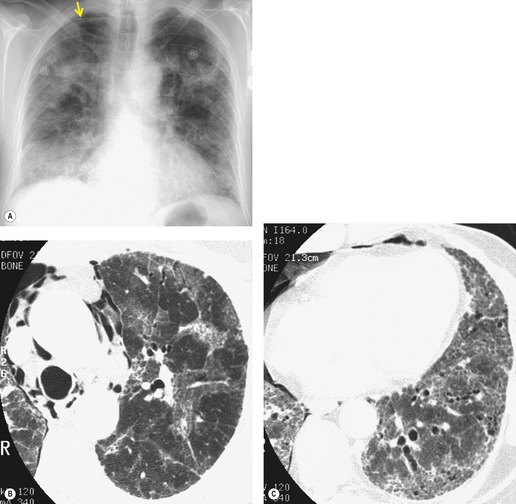
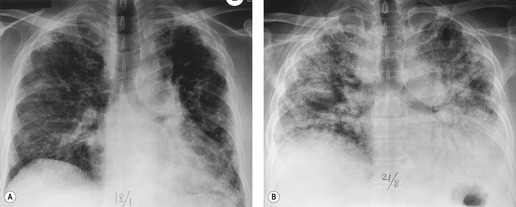
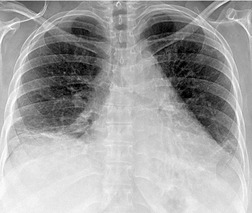
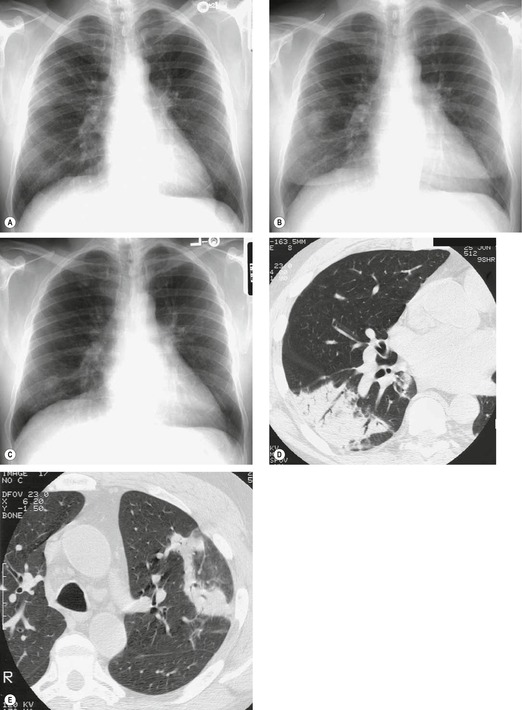
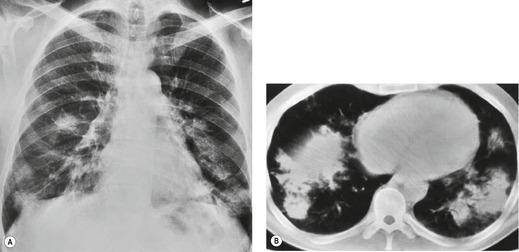
Almost all patients with UIP pattern due to IPF have abnormal chest radiographs when they present with symptoms. Indeed, when prior chest radiographs are available, basal reticular opacities are usually visible in retrospect for several years prior to the development of symptoms (Fig. 10.1). These presymptomatic opacities are commonly reported by the radiologist but, in the absence of symptoms or other abnormalities, disregarded by the clinician. Since early detection of IPF may be of value in treatment, the radiologist should emphasize the importance of these abnormalities, and ensure that they are investigated by careful review of the patient’s symptoms and signs, and, if appropriate, physiologic and HRCT evaluation. The classical chest radiographic appearance in patients with UIP is of basal reticulonodular pattern which becomes a coarser reticular, or honeycomb, pattern as the disease progresses (Fig. 10.2). The characteristic subpleural distribution of the abnormality may often be recognized in the mid- and upper lungs, along the lateral chest wall. The lung volumes usually decrease with time, particularly in the lower lobes, but in patients with associated emphysema, the radiographic lung volumes may be normal or even increased (Fig. 10.3).
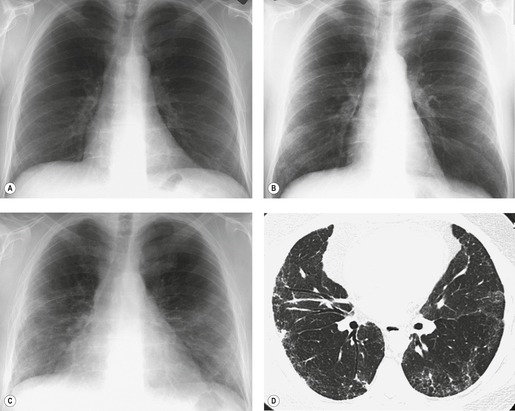
Pneumomediastinum (Fig. 10.2) is quite common in patients with UIP, and is often asymptomatic. Pneumothorax may be seen; while this may be small and asymptomatic, it may also be associated with a significant air leak if there is rupture of a honeycomb cyst. In patients with advanced UIP who develop pneumothorax, the stiffness of the lung often prevents marked lung collapse.
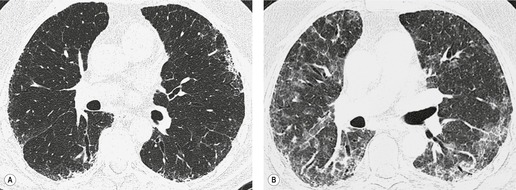
In about 50–70% of cases, UIP has a very characteristic appearance on CT. 79 The predominant CT pattern is usually either reticular, honeycombing, or a combination of both (Figs 10.3 and 10.4). 25 Traction bronchiectasis and/or bronchiolectasis is often seen. Although ground-glass abnormality is commonly present,25.80. and 81. it is usually associated with reticular abnormality and traction bronchiectasis, suggesting that it represents lung fibrosis rather than inflammation. Isolated ground-glass attenuation, if present, is usually sparse. Consolidation is uncommon in studies from North America and Europe, but appears to be more common in Asian series.79.80. and 81. The abnormalities are basal predominant in most, but may be diffuse. Peripheral, subpleural predominance is present in over 90%. The distribution of fibrosis may be asymmetric in up to 25% of cases, and asymmetry may be useful in distinguishing UIP from other fibrotic interstitial pneumonias. 80 In contrast to the homogeneous appearance of NSIP, the abnormalities of UIP often have a patchy distribution. As the disease progresses, it often appears to ‘creep’ up the periphery of the lung, causing subpleural reticular abnormality in the upper lungs (Fig. 10.4). In a study by Hunninghake et al., 82 this finding of subpleural lines in the upper lungs was useful in discriminating between UIP and other conditions.
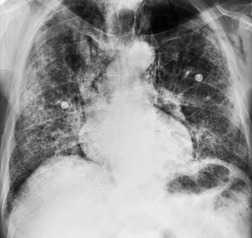
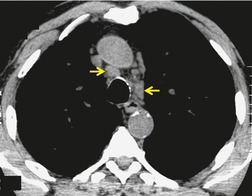
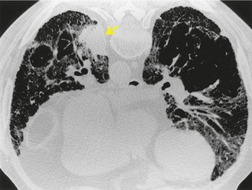
As with many other ILDs, mediastinal adenopathy may be seen on CT in UIP (Fig. 10.5). 83 The nodal enlargement typically involves only one or two nodal stations and the nodes usually measure less than 15 mm. The adenopathy is thought to be reactive. 84 It is never visible on the chest radiograph. The presence of lymph node enlargement does not appear to correlate with the extent of lung fibrosis, 83 but the number of enlarged nodes may relate to disease extent. 85 Pulmonary ossification, characterized by areas of punctate or linear calcification, may occur in areas of advanced fibrosis (Fig. 10.6), and will be better seen if the CT images are viewed at mediastinal windows. 86 Several other examples of the typical CT findings of UIP may be found in the section on collagen vascular diseases (see Figs 10.29, 10.31, 10.33 and 10.42 below).
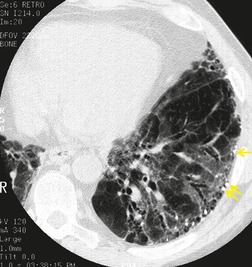
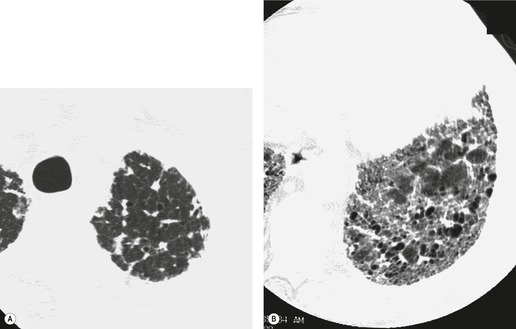
Numerous prospective and retrospective studies have shown that a confident or highly confident diagnosis of UIP, based on the CT features outlined above, has a specificity of over 90% for the pathologic diagnosis of UIP.25.87.88.89.90. and 91. Honeycombing in the lower lobes, and linear abnormality in the upper lobes, are the most reliable features for differentiating between UIP and its clinical mimics (Fig. 10.4). 82 In a study by Flaherty et al., 16 the observation of honeycombing on HRCT indicated the presence of UIP with a sensitivity of 90% and specificity of 86%.
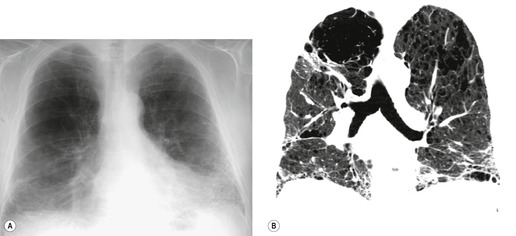
Because of the high degree of accuracy of HRCT in many cases of UIP, the diagnosis of UIP is commonly made based on clinical and imaging features, without the need for surgical biopsy. The ATS has published criteria for diagnosis of UIP in the absence of a surgical biopsy (Box 10.6). 18 These criteria indicate that ‘bibasilar reticular abnormalities with minimal ground glass opacities on HRCT scans’ are a major criterion for diagnosis of IPF. However, since publication of these criteria, the importance of honeycombing in the diagnosis of IPF has become more apparent,16. and 82. and a confident CT diagnosis of UIP is not usually made unless honeycombing is present. However, there is a substantial minority (30–50%) of cases of histologic UIP in whom a confident diagnosis of UIP cannot be made based on the CT appearances (Fig. 10.7).89.91.92. and 93. In these patients, the diagnosis of UIP can be made only by lung biopsy. A study by Flaherty et al. 68 suggested that the patients with histologic UIP who had definite or probable UIP by HRCT criteria had a shorter survival than those who had indeterminate HRCT findings. This is most likely because the typical HRCT criteria for diagnosis of UIP include the presence of honeycombing, and may therefore select patients with more advanced or more severe disease. This study reemphasizes the importance of seeking lung biopsy in patients in whom CT is not diagnostic of UIP. An equivocal, atypical, or nonspecific CT appearance should always prompt biopsy. Most importantly, the CT features must be interpreted in conjunction with a complete clinical evaluation.
Box 10.6
Major criteria
• Exclusion of other known causes of ILD such as certain drug toxicities, environmental exposures, and connective tissue diseases
• Abnormal pulmonary function studies that include evidence of restriction (reduced vital capacity often with an increased forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio) and impaired gas exchange (increased P(A-a)O2 with rest or exercise or decreased DLco)
• Bibasilar reticular abnormalities with minimal ground-glass opacities on HRCT
• Transbronchial lung biopsy or bronchoalveolar lavage (BAL) showing no features to support an alternative diagnosis
Minor criteria
• Age >50 years
• Insidious onset of otherwise unexplained dyspnea on exertion
• Duration of illness 3 months
• Bibasilar, inspiratory crackles (dry or ‘velcro’-type in quality)
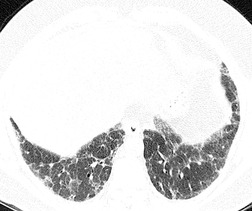
Initial reports of the CT features of IPF or fibrosing alveolitis suggested that the finding of ground-glass attenuation was associated with ‘alveolitis’ on biopsy, 94 which were potentially reversible on steroids and were associated with a favorable prognosis.22. and 95. However, the patients in those studies who had a large amount of ground-glass abnormality probably had NSIP or DIP rather than UIP. HRCT studies of ‘IPF’, ‘fibrosing alveolitis’, or ‘UIP’ published before 1995 are very difficult to interpret, because it is unclear how many cases of NSIP or DIP were included in these papers. As discussed above, under the current definition of UIP, alveolitis is not regarded as an important histologic component of the disease, and therefore ground-glass attenuation in UIP cannot be regarded as reflecting alveolitis. In contrast to DIP96 and NSIP, 97 true UIP shows inexorable progression and rarely if ever reverses in response to steroid treatment (Fig. 10.8). 96 When extensive ground-glass attenuation is present in patients with known UIP, the possibility of an acute exacerbation or accelerated deterioration should be considered.
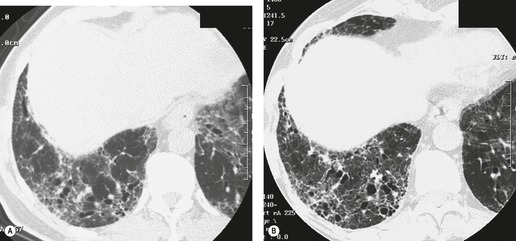
Several studies have evaluated sequential changes in IPF.24.81.98.99.100. and 101. Comparison of serial scans in patients with IPF depends on identifying comparable scan levels, by matching anatomic features such as vessels and bronchi. In untreated patients scanned at intervals of greater than 6 months, the extent of IPF increases progressively (Fig. 10.8). In patients who are treated, some areas of ground-glass attenuation progress to reticular abnormality and honeycombing, but some areas regress. Reticular abnormality usually progresses to honeycombing. 99 Areas of honeycombing increase inexorably in extent, 81 and the size of the honeycomb cysts also increases. 98 The presence or absence of honeycombing on baseline CT may impact disease progression. In a study by Jeong et al., 101 extent of disease decreased in five of 29 patients with UIP who did not have honeycombing on baseline CT, but in none of 22 with honeycombing. However, progression occurred in six of those without honeycombing, and in only two of those without honeycombing. The appropriate interval for detecting change in patients with IPF would appear to be about a year, though some patients may show clear evidence of change in 3–4 months. Of course, sequential scanning is indicated only if it will change management.
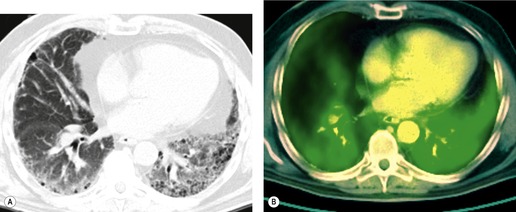
Positron emission tomography (PET) scans performed in individuals with lung fibrosis usually show increased metabolic activity in areas of lung fibrosis (Fig. 10.9); increased uptake of [18 F]2-fluoro-2-deoxy-D-glucose (FDG) was found in the fibrotic lungs in six of seven male patients with IPF. 102 One patient with end-stage lung fibrosis did not have increased uptake. In one of these patients the degree of uptake decreased after steroid treatment (when dyspnea decreased), and uptake increased again when steroids were tapered and symptoms increased. In a different study of 21 patients with a variety of infiltrative lung diseases, activity on FDG-PET did not help discriminate between IPF and non-IPF conditions. 103 There has also been interest in the use of 18F-proline as an agent to monitor fibrotic activity in the lung, but a recent study of seven patients with fibrotic lung disease studied with this agent showed low uptake in all patients with IPF. 104
Complications
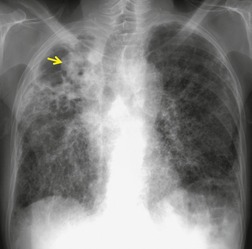
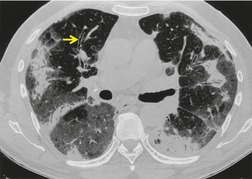
Important complications of IPF include infection, lung cancer, and accelerated deterioration (Box 10.7). 71 A variety of opportunistic infectious organisms may occur in treated patients with IPF, including Pneumocystis jirovecii (Fig. 10.10), Mycobacterium avium complex (Fig. 10.11), and mycetoma due to Aspergillus species or other organisms (Fig. 10.12).
Box 10.7
• Infection
– Pneumocystis jirovecii
– Nontuberculous mycobacteria
– Aspergillus
• Lung cancer
– Any cell type
• Acute exacerbation/accelerated deterioration
– AIP histology superimposed on background UIP
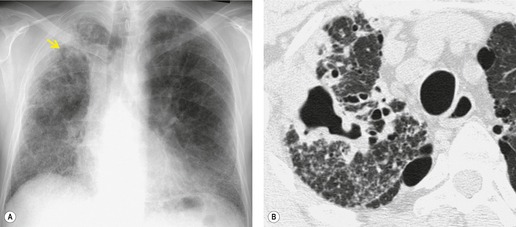
Lung cancer (Fig. 10.13) develops five times more frequently in patients with IPF than in the general population. 105 Lung cancer appears to be primarily associated with UIP, rather than with the other IIPs. It occurs most commonly in male smokers. 106 The prevalence of carcinoma in various series has ranged between 5% and 13%.65.71.107. and 108. Synchronous multiple lung cancers occur in up to 15% of patients with fibrosing alveolitis. 109 Adenocarcinoma and squamous carcinoma are the two most common cell types, with approximately equal prevalence in most series, and smaller numbers of patients with bronchioloalveolar cell carcinoma and small cell cancer.106. and 108. Lee et al. 108 found lung cancer in 32 (13%) of 244 cases of IPF seen over a 7.5-year period. The majority (55–65%) of these lung cancers occur peripherally, usually adjacent to honeycombing (Fig. 10.13), and most of the remainder are found in the upper lobes106. and 108. (Box 10.8).108. and 109. The cancers may appear as areas of poorly defined consolidation, 108 but are more commonly lobulated nodules. 106 The preferential occurrence of these lung cancers within areas of fibrosis may make them difficult to detect. Meticulous comparison of serial images is important, and evaluation for additional nodules is also important, since multiple synchronous cancers are relatively common. 109 The presence of lung fibrosis does not preclude resection for lung cancer, but the mortality and morbidity are higher than in those without lung cancer.110. and 111.
Box 10.8
• Occurs in areas of lung fibrosis
• Often multifocal
• Usually in patients with moderate or advanced lung fibrosis
Acute exacerbation of UIP
Between 10% and 25% of patients with IPF develop accelerated deterioration, or acute exacerbations, progressing to acute respiratory failure.70.112.113.114. and 115. Acute exacerbation is more acute than accelerated deterioration, but the precise distinction between these entities is unclear, and the terms are sometimes used interchangeably. The lungs of most of these patients have a histologic appearance of diffuse alveolar damage, similar to that of acute interstitial pneumonitis, though organizing pneumonia may also be seen, and has a better prognosis. Acute exacerbation is one of the most common causes of death in patients with IPF. 69
Accelerated deterioration or acute exacerbation of IPF presents with a relatively short onset of progressive dyspnea or cough, occasionally associated with systemic symptoms. There is usually a short prodrome of 4–8 weeks’ duration. Mortality in different series has varied from 20% to 100%, but exceeds 80% in most studies. 116 A recently published consensus definition for this entity requires the following: a previous or concurrent diagnosis of IPF, with unexplained worsening or development of dyspnea within 30 days, HRCT showing new bilateral ground-glass abnormality and/or consolidation superimposed on a background of usual interstitial pneumonia, and exclusion of other causes of decompensation including infection, left heart failure, and pulmonary embolism. 116 Precipitants for acute exacerbation may include thoracic biopsy or resection, chemotherapy, infection, and irradiation.117.118.119.120. and 121. Even patients with subclinical lung fibrosis evident on CT may be at risk for acute respiratory distress following thoracic resection surgery. 122 Acute exacerbation of IPF may be more common in Asian individuals (Box 10.9). 123
Box 10.9
• May occur in any fibrotic lung disease, but most common in UIP
• Precipitants may include surgery, drugs, infection
• Imaging shows ground-glass abnormality on background of fibrosis
• High mortality
The chest radiograph usually shows ground-glass abnormality or consolidation superimposed on background reticular abnormality (Fig. 10.14). On CT, acute exacerbation is characterized by diffuse, multifocal, or peripheral ground-glass attenuation (Fig. 10.15), 112 which must be distinguished clinically from opportunistic viral or pneumocystis infection. Consolidation may also be seen. In 17 patients with accelerated IPF, Akira et al. 112 classified the CT distribution of ground-glass attenuation as peripheral (adjacent to preexisting fibrosis), diffuse, or multifocal. Peripheral opacities were more likely to improve with corticosteroid treatment. On biopsy, the peripheral pattern of abnormality correlated with the presence of active fibroblastic foci in two patients, while the multifocal and diffuse patterns correlated with acute diffuse alveolar damage superimposed on UIP. On sequential evaluation of patients with acute exacerbation or accelerated deterioration, ground-glass attenuation was seen to evolve into consolidation with architectural distortion, and cystic lesions often developed. 113 In a subsequent study from the same group, evaluating 58 patients with acute exacerbation, the diffuse pattern of parenchymal abnormality, found in 34 patients, was associated with significantly worse survival than the multifocal and peripheral patterns (median survival 16 days, compared with 240 and 540 days respectively). 124
Nonspecific interstitial pneumonia
The key points about NSIP are:
• Despite the term ‘nonspecific’, the entity does in fact represent a specific set of histologic patterns, and the fibrotic subtype is often associated with a very typical CT appearance. 128
• There are two histologic subtypes, cellular and fibrotic, with the fibrotic subtype being more common.
It seems highly likely that, in the past, NSIP was categorized as IPF, UIP, or cellular interstitial pneumonia. The first description of this entity, in 1994, encompassed a group of 64 cases that had a histologic pattern which did not fit into any of the existing descriptions of interstitial pneumonia. 129 The histologic pattern of NSIP does not appear to be associated with a specific clinical syndrome, and the condition presents in a similar fashion to UIP. 3
The primary pathologic finding in NSIP3.4.128. and 131. is the presence of varying degrees of inflammation and fibrosis within alveolar walls (Box 10.10). In contrast to the patchy heterogeneity of UIP, these changes are uniform and diffuse within a given lung region. In the original series, cases were divided into three groups depending on the relative amounts of inflammation and fibrosis. 129 In 48% interstitial inflammation with lymphocytes and plasma cells was predominant (type 1); in 38% there was an approximately equal mix of inflammation and fibrosis with mature collagen and few fibroblasts (type 2); and in 14% there was predominant interstitial collagen with little inflammation and few fibroblasts (type 3). Most recent studies14.15.128. and 132. have divided NSIP into cellular and fibrotic subtypes, and have found a lower prevalence (5–25%) of the cellular pattern of NSIP, with a higher prevalence of the fibrotic type. In addition to the defining feature of alveolar wall thickening by inflammation or fibrosis, histologic findings may also include small areas of organizing pneumonia (50%), small fibroblastic foci (20%), macrophage accumulation in alveoli, and occasional lymphoid aggregates and granulomas.3. and 128.
Box 10.10
• Uniform expansion of alveolar septa
• Homogeneous appearance within microscopic field
• Variable amounts of inflammation and fibrosis
There is substantial interobserver variation in the histologic diagnosis of NSIP, and particularly in the criteria used to distinguish between NSIP, UIP, and organizing pneumonia. In a study by Flaherty et al. 17 three expert pathologists agreed on histologic diagnoses of NSIP or UIP in only 67% of cases, while Nicholson et al. 15 found that the kappa coefficient of agreement between two pathologists for distinguishing UIP from fibrotic NSIP was only 0.26. Additionally, patients with fibrosing ILD may have coexisting lesions of UIP and NSIP. A study by Flaherty et al. 17 of 109 patients who had biopsies of multiple lobes showed that discordant diagnoses of UIP and NSIP were obtained from different lobes in 28 (26%).
At presentation patients have a mean age of 45–55 years with a range of 9–81 years.12.128.129.133. and 134. The mean age at presentation is about 10 years younger than for UIP. 4 In contrast to UIP, NSIP is about twice as common in women as in men. 128 Although the majority of patients with NSIP are nonsmokers, 128 NSIP can in some cases be related to cigarette smoking. 135 Clinical features of NSIP resemble those of other IIPs, with progressive dyspnea and nonproductive cough preceding diagnosis by 6–12 months on average15.128.129.133. and 136. but ranging from weeks to years. 129 Systemic symptoms such as weight loss and fever may be present in up to 25% of cases, and in one series 21% of patients had clubbing. 12 NSIP usually responds to steroid treatment and the prognosis is considerably better than that of UIP. The 10-year survival rate in NSIP was 60% versus 10% in UIP in one series12 and this relatively favorable prognosis is confirmed by numerous other studies.14.15.16. and 137. Histologic changes correlate with outcome, a predominant cellular pattern having a better prognosis than a pattern that is largely fibrotic.129. and 137. Indeed, the long-term survival of patients with purely cellular NSIP is close to 100% in several series.14. and 15. However, the survival of patients with predominantly fibrotic NSIP is poorer, with 5-year survival ranging from 45% to 90%,14. and 15. and 10-year survival as low as 35%. 14
In contrast to UIP, which is usually idiopathic, NSIP is commonly associated with an underlying cause. In the original series of patients with NSIP, 129 46% had possible precipitating events or associated disorders (Box 10.11). These consisted of: connective tissue disorders in 16% (rheumatoid arthritis [RA], dermato/polymyositis, systemic lupus erythematosus [SLE], systemic sclerosis, SjS); other autoimmune disorders in 5%, including primary biliary cirrhosis and Hashimoto thyroiditis; recent surgery, pneumonia, or acute respiratory distress syndrome (ARDS) in 8%; and inhalation of organic antigens in 17%. In a more recent study, Kinder et al. 138 found that 15/17 cases classified as idiopathic NSIP at their institution met criteria for undifferentiated connective tissue disease. As discussed below, NSIP is the commonest histologic pattern identified in patients with scleroderma. Thus, connective tissue disease, drug toxicity, and hypersensitivity pneumonitis must always be considered in the differential diagnosis of a CT pattern suggestive of NSIP. Connective tissue disease should be suspected on CT if the patient has associated findings such as enlarged pulmonary arteries, pleural or pericardial effusion, or dilated esophagus. 139 Hypersensitivity pneumonitis should be considered if there are micronodules or areas of lobular mosaic attenuation or air-trapping. 140
Box 10.11
• Collagen vascular disease and other autoimmune diseases
• Hypersensitivity pneumonitis
• Drugs
• Inhalational exposures
Imaging appearances
The chest radiograph12.129.136. and 137. is abnormal in more than 90% of patients with NSIP, but changes are not well characterized in the literature. Abnormalities are usually bilateral with a basal predominance. They may be consolidative and patchy, reticulonodular, or mixed. Lower lobe volume loss is common, and traction bronchiectasis is frequently present (Fig. 10.16).
Published descriptions of the CT appearances of NSIP have varied quite widely,13.25.97.132.136.141.142.143. and 144. probably because of differing pathologic diagnostic criteria. However, a multidisciplinary workshop identified a relatively typical CT appearance among 67 patients who received a clinical–imaging–pathologic consensus diagnosis of NSIP (Box 10.12). 128
Box 10.12
• Lower lung predominance
• Peribronchovascular distribution with subpleural sparing
• Ground-glass abnormality often dominant
• Reticular abnormality with traction bronchiectasis
• ± Consolidation
• Honeycombing sparse or absent
Ground-glass pattern is the most common CT abnormality in all series of patients with NSIP, being found in 90–100% of patients.81. and 145. It is usually confluent, bilateral, and symmetric, with basal predominance (Fig. 10.17). It is commonly peribronchovascular in predominant distribution, 141 sometimes with sparing of the subpleural lung. 128 The peribronchovascular distribution and subpleural sparing may be helpful in distinguishing NSIP from UIP. Ground-glass opacity occurs as an isolated finding in about a third of patients, but is more commonly associated with other findings such as reticular abnormality or consolidation. Consolidation is seen in 0%132 to 98%144 of patients, almost always combined with ground-glass opacity. The wide variation in prevalence of consolidation may be due to the fact that some patients with NSIP have considerable amounts of histologic organizing pneumonia, so that it can be difficult for the pathologist to decide whether to classify an individual case as NSIP or organizing pneumonia. 4 Criteria for distinguishing NSIP from organizing pneumonia remain poorly defined. Like ground-glass opacity, consolidation is bilateral, basal and subpleurally predominant or peribronchovascular. Irregular linear or reticular opacities (Fig. 10.17) occur in about 75% of cases (range 29–91%).81.136.141.142. and 145. Traction bronchiectasis and bronchiolectasis is a characteristic feature, present in areas of ground-glass attenuation or reticular abnormality in 70–90% of cases. 145 (For other examples of the typical CT findings of NSIP see the section on scleroderma [Figs 10.44 and 10.45].)
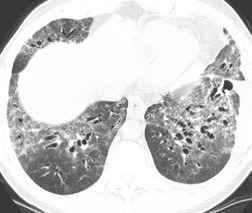
Honeycomb changes were recorded in 20–30% of patients in some series13.25.81.134.142. and 144. but were absent in others.97.136. and 141. This apparent discrepancy may be accounted for by interobserver variability in diagnosis of NSIP and UIP, and by the fact that histologic changes of UIP may coexist with those of NSIP. In general, honeycombing, if present in NSIP, is sparse. However when patients with NSIP are followed over time, honeycombing may increase in prevalence and extent. 81 Mildly enlarged mediastinal nodes are present in up to 80% of cases. 83 Pathologic/HRCT correlation141 has shown that ground-glass opacity is caused by cellular or fibrotic interstitial thickening; bronchial dilatation and irregular linear opacity indicates predominant fibrosis, and some areas of consolidation are caused by areas of organizing pneumonia.
Since the prognosis of cellular NSIP is better than that of the fibrotic type, it would be useful if CT were able to reliably discriminate between these entities. However, MacDonald et al. 132 suggested that there was substantial overlap between the CT findings of these subtypes, although cellular NSIP was less likely to be subpleural in distribution, and had a higher proportion of ground-glass type abnormality. Johkoh et al. 144 found that the extent of reticular abnormality and traction bronchiectasis correlated with increasing amounts of fibrosis on histologic grading. Honeycombing was seen mainly in patients with purely fibrotic NSIP. While traction bronchiectasis was present in almost all cases, it tended to involve more proximal airways (lobar and segmental bronchi) in patients with fibrotic NSIP. Patients with cellular NSIP were more likely to have pure ground-glass abnormality without reticular abnormality. However analysis of this issue is difficult because the proportion of patients with purely cellular NSIP is relatively small (six of 55 patients in the paper by Johkoh et al., 144 and five of 21 patients in the paper by MacDonald et al. 132).
Nishiyama et al. 97 evaluated the CT changes on follow-up of patients with NSIP after treatment. Parenchymal abnormalities showed improvement or resolution in 12 of 14 patients. Reticular abnormality, traction bronchiectasis, and ground-glass abnormality were all completely or partially reversible. One patient progressed, with ground-glass abnormality being replaced by honeycombing. In a similar study by Kim et al., 141 the extent of ground-glass abnormality decreased substantially on follow-up, but the extent of reticular abnormality decreased only slightly. In a serial CT evaluation of 24 subjects with NSIP, extent of lung fibrosis increased in nine, decreased in three, and was unchanged in 12. 101 A more long-term serial study of 23 patients with NSIP, with median follow-up of 61 months, found that the extent of ground-glass abnormality decreased over time, but tended to be replaced by increased fibrotic abnormality, with increased prevalence and extent of honeycombing, and development of a pattern suggestive of UIP in five patients. 81
Desquamative interstitial pneumonia, respiratory bronchiolitis interstitial lung disease
DIP and RB-ILD are important differential diagnostic considerations in cigarette-smoking patients presenting with the clinical syndrome of IIP. Because they are smoking-related, they are considered in the chapter on inhalational lung disease (see Chapter 8, pp 451–453).
Organizing pneumonia
Histologically, the organizing pneumonia pattern is characterized by the presence of intraluminal organizing fibrosis in the distal airspaces (bronchioles, alveolar ducts, and alveoli). 4 The distribution is usually patchy, and lung architecture is usually preserved. The connective tissue is characteristically all of the same age. There may be mild interstitial chronic inflammation. This pattern may be seen as the lung recovers from a variety of types of injury (Box 10.13). For this reason, a diagnosis of organizing pneumonia may sometimes be misleading if the biopsy is nonrepresentative (e.g. transbronchial biopsy) in patients with an organizing pneumonia response.
Box 10.13
• Organizing pneumonia as a major finding
• COP
• Secondary organizing pneumonia (collagen vascular disease, drugs)
• Focal organizing pneumonia
• Organizing pneumonia as a component of other pathologic entities
• Nonspecific interstitial pneumonia
• Hypersensitivity pneumonitis
• Eosinophilic lung disease
• Organizing diffuse alveolar damage
• Organizing infections
• Postobstructive pneumonia
• Aspiration pneumonia
• Fume and toxic exposures
• Radiation pneumonitis
• Organizing pneumonia as a reparative reaction around other processes
• Wegener granulomatosis
• Neoplasms
• Infarcts
Cryptogenic organizing pneumonia
COP was recognized as a distinct clinicopathologic entity in the 1980s.7.8. and 146. In the USA and Canada, it was called BOOP,8.147.148. and 149. or proliferative bronchiolitis, 150 because of the tendency of plugs of organizing pneumonia to occlude the smaller bronchioles. For this reason, it was often misleadingly classified as a small airways disease. However, its clinical, imaging, and pathologic features are distinct from those of small airways disease. There is some controversy about its inclusion as an IIP, 131 because the areas of organizing pneumonia involve the pulmonary airspaces rather than the interstitium, its clinical presentation is more subacute than UIP, DIP or NSIP, and its imaging features are those of airspace disease. However, it is included in the ATS/ERS classification of IIPs, 4 because of its idiopathic nature, its association with collagen vascular diseases, and its tendency to overlap with the other interstitial pneumonias, particularly NSIP.
COP is a rare condition, with a prevalence of 12 per 100 000 hospital admissions in a Canadian series, 151 and a reported incidence of 1.1/100 000 in Iceland. 152 Mean patient age is about 58 years (range 20–80 years),153. and 154. and sex incidence is equal. 151 Twenty-five to fifty per cent of patients give a history of an influenzalike prodrome followed by an illness lasting between a week and several months, characterized by cough (persistent and commonly nonproductive), exertional dyspnea, malaise, fever, and weight loss. Less common complaints include pleuritic pain147. and 155. and hemoptysis.155.156. and 157. Given these symptoms, and radiographic findings of airspace opacity, it is not surprising that 50% of patients are initially diagnosed as having infective pneumonia. 151 On clinical examination of the lungs, fine, dry crepitations occur in 70–90% of patients; clubbing is rare. 158 The erythrocyte sedimentation rate and C-reactive protein level are usually raised and may be very high.156. and 159.
Pulmonary function tests show a restrictive abnormality, commonly with a diffusion defect and hypoxemia at rest or on exertion.159. and 160. Occasionally there are obstructive features. 151 Bronchoalveolar lavage is nonspecific and shows mixed cellularity usually with predominant lymphocytes accompanied by neutrophils and eosinophils with or without foam cells, mast cells, and plasma cells. 160 Neutrophil/eosinophil predominance indicates an unfavorable prognosis. 161 The histologic diagnosis may be made by transbronchial biopsy162 in up to two-thirds of patients151. and 163. especially if a step sectioning technique is used. 159 CT may be helpful in identifying a suitable site for transbronchial biopsy. Bronchoscopy may also be important to exclude other causes of consolidation such as infection, aspiration, or bronchioloalveolar cell carcinoma. In cases where there is diagnostic doubt, thoracoscopic biopsy will be necessary.
Imaging findings
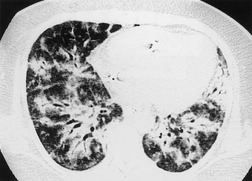
The imaging hallmark of COP is bilateral, patchy, airspace opacity (Fig. 10.18), with a density which ranges from consolidation to ground-glass opacity (Box 10.14). Airspace opacity is usually bilateral (70–90%) and asymmetric. In about 10% of cases, opacities are unilateral or focal.59.156.164. and 165. Consolidation, seen in about 80% of cases, is nonsegmental and commonly 2–6 cm in diameter. It generally shows no craniocaudal predilection though some series have shown a basal predominance.8.158. and 166. On CT, the consolidation often has a peribronchovascular2 or subpleural distribution and the subpleural distribution may be apparent on the chest radiograph.151.155.162. and 164. The consolidation commonly contains air bronchograms (Fig. 10.19) with dilated airways. Consolidation tends to migrate (Box 10.15; Fig. 10.18) and to come and go even without treatment.7.155.159.166.167.168. and 169. The consolidation may uncommonly cavitate.156.170. and 171. A perilobular pattern (poorly defined opacity along interlobular septa) (Fig. 10.20) was found in over 50% of patients with COP in one study and may help suggest the diagnosis. 172 The reverse halo or atoll sign (ringlike opacity with central ground-glass abnormality), found in six of 31 subjects in one series, 173 may also be helpful in suggesting the diagnosis, 174 although this sign may also be encountered in other conditions such as vasculitis, pulmonary infarction, or fungal infection.
Box 10.14
• Lower lung predominance
• Peripheral and/or peribronchovascular distribution
• Patchy consolidation
• Ground-glass abnormality
• Other patterns
– Nodular
– Bandlike
– Perilobular
– Reverse halo
Box 10.15
• Organizing pneumonia
• Eosinophilic lung disease
• Simple eosinophilic pneumonia (Löffler syndrome)
• Chronic eosinophilic pneumonia
• Drug/parasite hypersensitivity
• Allergic bronchopulmonary aspergillosis
• Churg–Strauss syndrome
• Diffuse alveolar hemorrhage
• Recurrent pulmonary infarction
• Recurrent aspiration
• Vasculitis
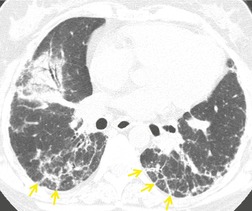
About 60% of patients175 have ground-glass opacity on CT (Fig. 10.21), often with a lobular, mosaic distribution.176. and 177. About 30–50% of patients demonstrate nodules either as an isolated finding or more commonly as part of a mixed pattern with consolidation (Fig. 10.22). Nodules show a great range in size but are most commonly in the 1–10 mm range, smooth, well defined and sometimes with a centrilobular or peribronchovascular distribution. 164 They are usually bilateral and grossly distributed in a random fashion. In a small number of patients some nodules are larger (10–20 mm) and may contain air bronchograms or have ill-defined margins (‘airspace’ nodules). Other findings described in a minority of patients include: peripheral reticulation and irregular lines, particularly in the lower zones;147.151. and 178. bronchial dilatation and wall thickening (Fig. 10.21); 164 large nodular or masslike peripheral opacities; 147 and satellite lesions; 179 bandlike opacities with central air bronchograms; 174 mild lymph node enlargement in 14–42% of patients;84.147. and 164. and small pleural effusions, either bilateral or unilateral, in about 20–30% patients.151.164.180. and 181. The finding of reticular abnormality on the chest radiograph or CT in a patient with biopsy-proven organizing pneumonia suggests that the patient will not respond to treatment, and may develop progressive fibrosis, likely due to evolution to NSIP.156. and 178. Other examples of the typical CT findings of organizing pneumonia may be found in the section on collagen vascular diseases (see Figs 10.32 and 10.52 below, and Box 10.16). Lesions of organizing pneumonia may be hypermetabolic on PET imaging. 182 In a study of 22 patients with primary and secondary organizing pneumonia, consolidation was associated with mildly increased uptake of FDG on PET scanning (mean SUV 3.1), and the degree of activity correlated with cellular measures of disease activity on bronchoalveolar lavage. 183
COP occasionally resolves spontaneously8.160. and 184. but usually requires treatment with steroids. With therapy, clinical and radiographic signs clear in weeks in 30–60% of patients, but there is a substantial relapse rate (50% in two recent studies)185. and 186. and up to one-third of patients are left with persistent radiographic abnormality. 154 A minority of patients with COP progress, despite treatment, to lung fibrosis with honeycombing.156. and 170. Progression is more common in those with longer duration of symptoms and those who have linear radiographic abnormalities on initial chest radiograph or reticular abnormality on CT.156. and 178. It is possible that some of these patients may have NSIP or UIP, with a nonrepresentative biopsy sample showing predominant organizing pneumonia.
There is a 3–13% mortality in various series, many of which combine cases of primary and secondary OP43. and 161. and generally the prognosis is less good in organizing pneumonia associated with connective tissue disorders.154.161. and 180. A subgroup of patients with cryptogenic or secondary organizing pneumonia is described with a fulminant course and high mortality.187.188.189. and 190. Histologic examination of biopsies in these cases has shown organizing pneumonia together with UIP and diffuse alveolar damage. 161 This entity most likely represents a form of acute exacerbation of diffuse lung disease.
Secondary organizing pneumonia
Organizing pneumonia is commonly associated with a recognized precipitating cause and in the Mayo Clinic series of 74 patients, 36% were classified as secondary, compared with 50% cryptogenic cases. 180 These authors found no distinguishing clinical, imaging, or pathologic features180 between COP and secondary organizing pneumonia apart from a worse overall prognosis in the secondary group, as noted by others. 187 The poorer prognosis includes a slower and less complete resolution and a higher respiratory mortality.
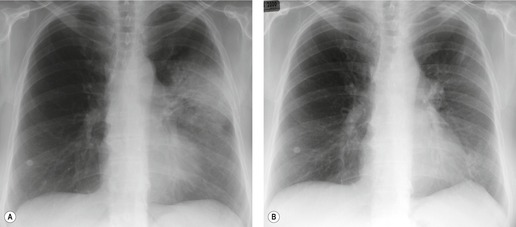
Associations and causes of secondary organizing pneumonia are listed in Box 10.16.4.151.160. and 161. In clinical practice, the common causes of an organizing pneumonia pattern are collagen vascular disease (particularly polymyositis/dermatomyositis, and RA), drugs, and lung or bone marrow transplantation. One interesting clinical entity is the occurrence in women of migratory organizing pneumonia, within a year following radiation therapy for breast cancer (Fig. 10.23).169.191.192.193. and 194. These patients may have typical symptoms of COP or may be asymptomatic, and show typical migratory pulmonary opacities, both within and outside the radiation port. They usually respond well to steroids, though with a high prevalence of relapse on withdrawal of steroids. It is thought that the radiation treatment in some way ‘primes’ the immune system of the lung for the development of organizing pneumonia. 192
Focal organizing pneumonia
Focal organizing pneumonia, presenting with a single discrete focal nodule, mass, or consolidation on chest imaging, is an uncommon presentation of organizing pneumonia, usually not associated with the clinical syndrome of COP. 180 Lung resection is commonly performed because of suspicion of malignancy. Maldonado et al. 218described 26 cases, identified as solitary nodules (6–68 mm diameter). About 40% of patients had symptoms, most commonly cough. Four patients had symptoms suggestive of an antecedent respiratory infection, and a further three had infection identified at biopsy. Etiologies identified in other reports have included chemotherapy with bleomycin. 219 Underlying COPD may be present. 218 Kohno et al. 220 subdivided the CT appearances in 18 subjects into three types: small irregular masses often with pleural tags, all less than 2 cm diameter; larger oval lesions with broad pleural contact, some with satellite nodules, air bronchograms, and converging peripheral lung vessels; and oval lesions lying along bronchovascular bundles, again sometimes accompanied by pleural tags and satellites. Overall 94% of lesions had irregular margins and about 50% of lesions had air bronchograms, satellites, or pleural tags. Follow-up CT examinations showed partial or complete resolution, and the authors stressed the usefulness of such examinations in management. Cavitation may occur.170. and 221. In the report by Maldonado et al., 218 all four lesions studied by FDG-PET were hypermetabolic, and all seven studied with dynamic enhancement CT showed enhancement by more than 15 HU. Focal organizing pneumonia may also present with a ground-glass attenuation nodule, and, in a recent series, organizing pneumonia/NSIP accounted for 10 of 49 subjects with such nodules who underwent surgical biopsy. 222
Acute interstitial pneumonia
AIP was first described in 1944 by Hamman and Rich, 223 who called it acute diffuse interstitial fibrosis of the lungs. It proved fatal in all four of their subjects. Despite this early description, AIP was not identified as a distinct form of IIP until the histopathologic description of cases in 1986. 224 The pathology is that of organizing diffuse alveolar damage (DAD) and the condition resembles ARDS in all respects save for the lack of an identifiable precipitating event. Synonyms include Hamman–Rich syndrome, accelerated interstitial pneumonitis, and idiopathic ARDS. Key features of the condition are: fulminant respiratory failure in a previously healthy individual, without an identifiable precipitating event, pathologic changes of organizing DAD, imaging changes of ARDS, and a high mortality, in the order of 80–90%.
AIP may be differentiated histologically into acute, organizing, and fibrotic phases. In the acute exudative phase, there is diffuse alveolar interstitial thickening due to edema, and associated hyaline membranes. The histologic lung changes of organizing DAD224.225. and 226. consist of fibroblast proliferation in the alveolar interstitium. Collagen production is also usually mild.224. and 226. Hyaline membranes are usually more sparse. 154 Changes are diffuse and temporally uniform. 224 Other findings include hyaline membranes, intraalveolar organization, type II pneumocyte proliferation, and thrombi in small vessels. 227 In the fibrotic phase, alveolar wall collapse and apposition occurs. 154 Known causes of DAD (Box 10.17) must of course be excluded.
Box 10.17
• Infection
• Immunocompromise/transplant
• Acute exacerbation of fibrotic lung disease
• Collagen vascular disease
• Drug toxicity
• Toxic inhalation
• Sepsis
• Transfusion-related acute lung injury
• Shock
• Trauma
In a review of 58 patients who received a diagnosis of DAD on surgical lung biopsy over a 7-year period, the most common causes were infection and complications of transplantation. 228 AIP (i.e. DAD with no identified cause) accounted for 21% of these cases. Acute exacerbation of IPF (12%) and connective tissue diseases (16%) were also important causes. Hospital mortality was highest for those with acute exacerbation of IPF (86%), and mortality for AIP and connective tissue disease was 50% and 56% respectively. In a similar study of 36 patients undergoing mechanical ventilation who underwent open lung biopsy for diffuse pulmonary infiltrates of unknown etiology, IIP (AIP, exacerbation of UIP, COP, and NSIP) accounted for the majority of cases in which a diagnosis was made. 229
Clinically, previously healthy patients develop rapidly increasing dyspnea with fever over a few days, often following a short influenzalike prodrome. Males and females are equally affected, with a wide age range (13–83 years); mean ages also vary widely in various series from 28 to 65 years.230. and 231. Fulminant respiratory failure rapidly follows, necessitating ventilatory support. Steroid treatment is usually tried but its efficacy is unclear. In five series comprising 64 patients,223.224.226.230. and 231. mortality was 78%.
The radiographic appearances of AIP are similar to those of ARDS. The chest radiograph shows diffuse consolidation, sometimes with a zonal predominance and often with air bronchograms. 230 The distribution is often patchy (Fig. 10.24), sparing the costophrenic sulci. Lung volumes are usually low. As the disease progresses, pulmonary consolidation increases. As the DAD moves from the exudative to the organizing stage, the radiograph tends to show less consolidation, evolving to a ground-glass appearance with irregular linear opacities. The radiographic appearances of AIP, like those of ARDS, may be substantially affected by ventilator settings, with increasing airway pressures resulting in an apparent decrease in consolidation and increase in ground-glass abnormality.
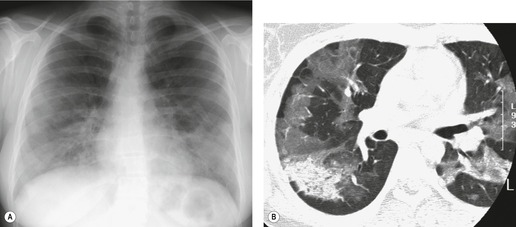
On HRCT113.230.231.232.233.234.235. and 236. (Fig. 10.24), ground-glass opacity is a universal finding. It is commonly diffuse and patchy, sometimes with a geographic pattern. The second commonest HRCT finding, in 70–90% of patients, is consolidation, often most evident in the dependent lung. In 30–70% of patients, septal thickening is found in areas of ground-glass opacity, perhaps corresponding to alveolar collapse adjacent to septa. Honeycombing is seen in 12–26% of cases, and should lead to suspicion of background fibrosing interstitial pneumonia. Pleural effusions were found in 38% of patients in one series of patients with AIP. 235 Effusions, if present, are usually small.
The CT features of AIP (Box 10.18) are similar but not identical to those of ARDS237. When Tomiyama et al. 236 compared the CT features of AIP with those of ARDS, the main differences were a higher prevalence of honeycombing and lower prevalence of septal thickening in AIP. The abnormalities of AIP were more likely to be symmetric, and to have lower lung predominance, than those of ARDS. The cases of AIP with honeycombing are presumed to represent an acute exacerbation of previously unrecognized UIP.
Box 10.18
• Diffuse distribution with lobular sparing
• Consolidation
• Ground-glass abnormality
• Traction bronchiectasis (organizing phase)
• ± Honeycombing
Ichikado et al. 231 described a close correlation between the CT findings and the phase of AIP. The acute phase is characterized by ground-glass attenuation and consolidation. The presence of traction bronchiectasis suggests that the disease is in the organizing phase, while honeycombing is seen in the fibrotic phase. When the CT features of patients with AIP who survived were compared with those who died, architectural distortion was more commonly seen in the nonsurvivors. 232 However, because of overlap in CT findings between survivors and nonsurvivors, this observation is of limited value in individual cases. 238
Lymphoid interstitial pneumonia
LIP is histologically characterized by marked infiltration of the interstitial space, as well as to a lesser extent the alveolar space, by monotonous sheets of lymphocytes. Diseases associated with LIP include dysproteinemias (such as common variable immunodeficiency), autoimmune disorders (particularly SjS), autologous bone marrow transplantation, and viral, mycobacterial, and HIV infections. HIV-related LIP occurs much more commonly in children than in adults. 239 Intrathoracic Castleman disease is commonly associated with LIP. 240 There are also familial and idiopathic forms of LIP.
In the classification of ILD, LIP has been classified either as an interstitial pneumonia or as a lymphoproliferative disorder. It is thought to represent a reactive lymphoid hyperplasia; evolution to frank lymphoproliferative disease is now thought not to occur.4.241. and 242. Though not usually idiopathic, it was included in the ATS/ERS classification of IIPs4 largely because it is important in the histologic differential diagnosis of interstitial pneumonia.
LIP is substantially more common in women, and most commonly presents in the fourth to seventh decades. Dyspnea and a nonproductive cough are common and may be present for months to years. Constitutional symptoms of fever, weight loss, and arthralgia may occur. There may be symptoms of a well-defined or evolving connective tissue disease. On examination inspiratory dry crackles are common, while clubbing occasionally occurs. Dysproteinemia may be present. On physiology, a restrictive ventilatory defect, with preserved airflow, is found. The diagnosis of LIP requires histologic confirmation, usually by a surgical biopsy. Immunologic techniques are often required to separate LIP from a well-differentiated pulmonary lymphoma.
On the chest radiograph LIP is generally characterized by basal predominant ground-glass or reticular abnormality (Fig. 10.25). 239 In a study of the radiographs of 16 patients with LIP related to HIV infection, Oldham et al. 243 found a fine reticular or reticulonodular pattern in five patients, coarse reticulonodular infiltrates in two, and reticular or reticulonodular opacities with superimposed patchy alveolar infiltrates in nine.
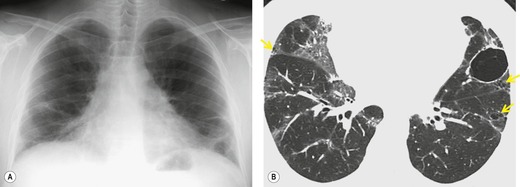
Johkoh et al. 244 described the CT features of LIP in a series of 22 patients (Box 10.19). Ten of these patients had SjS, seven had multicentric Castleman disease with hypergammaglobulinemia, two had AIDS, and three had no underlying disease. In this series the dominant CT findings, present in all patients, were ground-glass opacity (Fig. 10.25) and poorly defined centrilobular nodules. These findings were usually diffuse in distribution. Subpleural nodules, bronchovascular thickening, and septal thickening were also common, seen in 19 of 22 patients. Discrete cystic airspaces ranging from 1 mm to 30 mm were found in 15 patients (see Fig. 10.25).245.246.247. and 248. They differ from honeycomb cysts in that they are not clustered, and may be peribronchovascular. Mediastinal or hilar lymphadenopathy was seen in 15 patients, particularly in those with intrathoracic Castleman syndrome. Areas of consolidation were found in nine patients. Other, smaller, series have confirmed that ground-glass abnormality, cysts, and nodules are the salient CT features of LIP.249. and 250. The centrilobular nodules appear to correlate with peribronchiolar lymphocytic infiltration, while the ground-glass abnormality and consolidation presumably correlate with more diffuse infiltration of the alveolar interstitium by lymphocytes. The cysts appear to represent dilated small bronchi and bronchioles caused by lymphocytic infiltration and partial obstruction.245. and 248. On follow-up CT, the ground-glass abnormality and centrilobular nodules of LIP generally improve on treatment, but new cysts may develop in areas previously occupied by centrilobular nodules, while areas of consolidation may evolve into honeycombing. 251
Box 10.19
• Ground-glass abnormality
• Nodules (centrilobular or subpleural)
• Cysts
Honda et al. 247 compared the CT features of LIP with those of lymphoma, and found that large nodules (11–30 mm in diameter), consolidation, and pleural effusions were much more common in lymphoma, while cysts were much more common in LIP. Several other examples of the typical CT findings of LIP may be found in the section on SjS (see Figs 10.54 and 10.55 below).
Familial lung fibrosis and surfactant deficiencies
About 5% of cases of lung fibrosis have a family history of fibrosis in a family member. 252 Telomerase deficiency was identified in 8% of cases of familial lung fibrosis, 253 and telomere shortening has been identified in 25% of sporadic and 37% of familial pulmonary fibrosis. 254
A study of 713 individuals from 111 families with familial fibrosis found 309 with definite or probable lung fibrosis. 255 Genetic analysis suggested autosomal dominant inheritance with reduced penetrance. Among siblings in this cohort, cigarette smoking was associated with an odds ratio of 3.6 for presence of disease. 255 UIP was by far the most common pattern, found in 80% of individuals, followed by NSIP (6%), and organizing pneumonia (1%), with about 12% of cases not conforming to a recognized phenotype. About 45% of familles demonstrated different phenotypes (UIP, NSIP, organizing pneumonia) within the same family, suggesting that various subtypes of IIP may share a common pathogenesis. In a study of 143 asymptomatic family members of patients with familial IPF, CT abnormalities suggesting early ILD were identified in 31 (22%). 256 The most common HRCT findings in these subjects were increased septal lines, peribronchovascular thickening, reticulation, and ground-glass opacities. Subpleural cysts and honeycombing were observed in some subjects. In a CT study of nine subjects with familial pulmonary fibrosis, the findings were similar to those of UIP, except for a lower prevalence of honeycombing (present in only three patients), and a higher prevalence of upper lobe predominance (found in two patients). 257 Progression occurred in eight of nine subjects after a median interval of 49 months.
Pulmonary surfactant is a phospholipid–protein complex synthesized in alveolar type II cells and necessary for maintaining alveolar expansion at end-expiration. 258 Mutations in three genes of the surfactant synthetic pathway, surfactant proteins-B and -C (SFTPB and SFTPC) and the ATP-Binding Cassette member A3 (ABCA3), disrupt surfactant function and result in accumulation of surfactant in the lung parenchyma. Recognition of these deficiencies has been an important advance in the understanding of ILD in childhood. A review of the clinical and imaging features in nine patients with ABCA deficiency indicated that most presented as neonates, although presentation could be delayed up to age 4. 259 Ground-glass opacity was present on CT in all cases. All patients imaged beyond infancy developed fine or coarse peripheral interstitial septal thickening. Five patients eventually developed small cysts. Hilar and mediastinal lymphadenopathy, consolidation, atelectasis, pleural thickening and air-trapping were also seen. Histologic evaluation in children who had repeated evaluation showed evolution from an initial pattern suggestive of pulmonary alveolar proteinosis to a later pattern suggestive of NSIP. Histologic DIP has also been described in adolescents with ABCA deficiency. 260
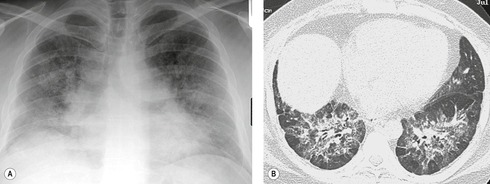
SFTPB deficiency is a rare entity, which usually presents with neonatal respiratory distress. 261 Histology most commonly shows congenital pulmonary alveolar proteinosis, but may also show DIP. In two affected neonates, the features were characterized by ground-glass and reticular abnormality with interlobular septal thickening. 262 SFTPC deficiency is more common than SFTPB deficiency, and the clinical presentations and outcomes of lung disease associated with SFTPC mutations are considerably more variable, even within families known to carry a mutation. 261 Inheritance is autosomal dominant. Some individuals with SFTPC deficiency are asymptomatic. Affected individuals may present with neonatal respiratory distress, or may present in childhood or adulthood with gradual onset of dyspnea. Identified histologic patterns have included UIP and NSIP. 263 Surfactant protein C (SPC) deficiency may account for some cases of familial fibrosis in both children and adults (Fig. 10.26),263. and 264. but the prevalence of the SFTPC gene is low in sporadic lung fibrosis. 265
Diagnostic accuracy of CT in IIP
UIP
As a result of the marked differences in prognosis and in management, the most important diagnostic distinction is between UIP and other entities. Table 10.2 indicates the CT features which help make this distinction. In a study of 91 patients presenting with a clinical syndrome suggestive of IPF, 60% had UIP on biopsy, 13% had respiratory bronchiolitis, 8% had hypersensitivity pneumonitis, 7% had NSIP, 3% had sarcoidosis, while the remaining patients had a variety of diagnoses including emphysema, bronchioloalveolar cell carcinoma, organizing pneumonia, eosinophilic pneumonia, pulmonary hypertension, silicosis, and Langerhans histiocytosis. 82 In this study, 91 as in many others,25.87.88.89.90. and 266. a confident CT diagnosis of UIP was correct in over 90% of cases. Most recently, in a multicenter treatment study, a ‘highly suggestive’ CT diagnosis of UIP, made in 49% of readings, was correct in 92% of cases. 267 Weighted kappa for expert radiologists determining presence or absence of UIP in this study was 0.40.
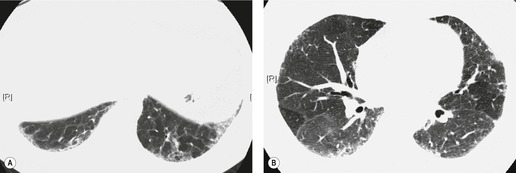
It is clear that honeycombing is the most important diagnostic feature of UIP, particularly in making the distinction between UIP and NSIP.16. and 82. Sumikawa et al. 79 found that the extent of honeycombing was the strongest predictive feature for distinguishing between UIP and fibrotic NSIP. The presence of upper lobe irregular lines has also been found to be another independent discriminator between UIP and non-UIP. 82 Chronic hypersensitivity pneumonitis (CHP) is an important aspect of the differential diagnosis of NSIP and UIP (see Chapter 8, p 461). Features that favor CHP over NSIP or UIP include upper- or mid-lung predominance, the presence of lobular decreased attenuation and air-trapping (Fig. 10.27), and the presence of poorly defined centrilobular nodules.268. and 269. However, CHP may present with a histologic pattern identical to NSIP129 or less commonly UIP. 270 Therefore, a thorough clinical evaluation for possible sources of hypersensitivity pneumonitis is important in all patients presenting with CT features suggestive of UIP or NSIP.
Pulmonary sarcoidosis, with its reputation as a great mimic, may rarely simulate the basal and peripheral pattern of UIP, with subpleural cysts being found in a small number of cases. 271 However, the cysts of sarcoidosis are often larger than those of UIP.
NSIP, DIP and organizing pneumonia
Early studies of the CT differential diagnosis of NSIP noted difficulty in distinguishing it from other ILDs.25. and 134. However, since these studies, our understanding of the CT findings of NSIP has evolved, and the typical pattern of basal predominant ground-glass attenuation and reticular abnormality with traction bronchiectasis has been recognized. The greatest challenge, both for pathologists and radiologists, is distinguishing between fibrotic NSIP and UIP. In a study by MacDonald et al. 132 of 21 patients with NSIP and 32 patients with UIP, the sensitivity, specificity and accuracy of a CT diagnosis of NSIP were 70%, 63%, and 66%, respectively. UIP was falsely diagnosed in 21 (33%) of 64 readings of patients with fibrotic NSIP. Conversely, NSIP was diagnosed in 48 (38%) of 128 readings in patients with histologic UIP. In a study by Elliot et al. 272 of 29 patients with NSIP and 24 with UIP, a predominant pattern of ground-glass attenuation and/or reticular opacity, with minimal to no honeycombing, was demonstrated in 57 (98%) of 58 readings in patients with NSIP, and in 21 (43%) of 48 readings of patients with UIP. Conversely, the presence of honeycombing as a predominant feature had a specificity of 98% and positive predictive value of 96% for UIP. The positive predictive value of a confident diagnosis of NSIP was 77%, while the positive predictive value of a confident diagnosis of UIP was 91%. In a more recent study that also included hypersensitivity pneumonitis, the positive predictive values for confident diagnoses of NSIP and UIP were 94% and 100%, respectively. 140 These studies indicate that it is possible to confidently identify UIP when honeycombing is present, but a substantial minority of patients with UIP have CT findings similar to those of NSIP, and can only be distinguished by biopsy. As indicated above, pathologists show substantial variation in differentiating between UIP and NSIP, so the gold standard for this diagnosis is not established, and the most accurate assessment probably requires clinical–imaging–pathologic correlation. 128 For example, if the surgical biopsy is interpreted as NSIP, and substantial honeycombing is present on CT, then the imaging findings trump the pathology findings, in terms of the patient’s prognosis if not the final diagnosis.
At the other end of the spectrum, it may be difficult to distinguish histologic NSIP from organizing pneumonia. Focal areas of organizing pneumonia are seen histologically in about 50% of cases of NSIP. 129 As with UIP, the amount of organizing pneumonia present on an individual biopsy specimen may vary with the site of biopsy. On CT, the main distinguishing features between organizing pneumonia and NSIP are that the consolidation of organizing pneumonia is more patchy and that reticular abnormality and traction bronchiectasis are less common. No clear borderline exists to delineate the extent of consolidation in organizing pneumonia from NSIP, and indeed there are cases where organizing pneumonia appears to evolve into NSIP. Organizing pneumonia should be favored in patients with patchy consolidation, while NSIP is favored in patients with basal predominant confluent abnormality associated with evidence of fibrosis.
The imaging features of NSIP may overlap with those of DIP. A subpleural distribution, and the presence of small cysts, should favor the diagnosis of DIP. However, the distinction between NSIP and DIP is not critical, since the treatment of these entities is similar. The long-term survival in DIP is excellent, and similar to that of cellular NSIP. 41
Acute interstitial pneumonia
In patients presenting with the syndrome of AIP, the other IIPs are not usually included in the differential diagnosis. However, the differential diagnosis must include infectious pneumonia, acute hypersensitivity pneumonitis, acute eosinophilic pneumonia, pulmonary hemorrhage, drug or transfusion reaction, and cardiac or noncardiac edema. Tomiyama et al. 235 compared the CT features of AIP with other causes of acute respiratory failure. In their study, the combination of airspace consolidation, centrilobular nodules, and segmental distribution was found in most cases of infectious pneumonia, but was not seen in AIP. In particular, the finding of centrilobular nodules or tree-in-bud pattern was very suggestive of infection. Traction bronchiectasis and honeycombing, rarely seen in the other acute pneumonias, should suggest AIP. The presence of septal thickening or pleural effusion as predominant findings should suggest acute eosinophilic pneumonia (see Chapter 11, p 660). In the study by Tomiyama et al., 235 a correct first-choice diagnosis of AIP was made in 90% of cases of AIP, based on the findings indicated above.
Interdisciplinary approach to diagnosis
There is moderate interobserver variation among radiologists and pathologists in characterization of diffuse lung disease. In a study of 131 patients evaluated by 11 thoracic radiologists, agreement on the first choice diagnosis was moderate (kappa 0.48). 273 In a similar study among pathologists, agreement was slightly lower (kappa 0.38, increasing to 0.43 when multiple biopsies were available). 274 Two studies by Flaherty et al.,275. and 276. using sequential presentation of clinical, imaging, and pathologic information in patients with suspected IIP, showed that interobserver variation regarding the first choice diagnosis improved at each stage of the process. Histopathologic information had the greatest impact on the final consensus diagnosis, but presentation of clinical and imaging information resulted in alteration of pathologic diagnosis in 11 of 58 patients. 276 Community clinicians and radiologists were more likely to assign a diagnosis of IPF than academic radiologists. 275 These studies make it clear that there are times when typical imaging or clinical findings (particularly typical CT features of UIP or chronic hypersensitivity pneumonitis) can overrule the pathologic diagnosis.
Correlation between structure and function in IIPs
The functional deficit in patients with lung fibrosis is typically restrictive with reduced compliance, lung volume, and decreased gas diffusing capacity. The pulmonary function indices that reflect these abnormalities are routinely used in clinical practice to make an assessment of the extent of disease and monitor progress. These tests are limited by inability to account for the regional inhomogeneity of fibrosing lung disease. However, the severity of physiologic impairment, 66 and the change in physiologic parameters over a 1-year interval, are good predictors of long-term survival.277. and 278. Most studies show substantial correlations between pulmonary function test data and visual assessment of disease extent by CT.32. and 279. However there is no widely accepted, reproducible, standardized scoring system for describing the visual extent of lung disease on CT.
Compared with chest radiographs, interobserver variation decreases substantially when the extent of parenchymal abnormality is estimated by CT.280. and 281. The most commonly used technique for the quantitation of disease extent is subjective visual estimation.20.32. and 281. However, some of the more subtle abnormalities caused by interstitial fibrosis may escape visual detection, and more sophisticated approaches to the quantification of ILD include analysis of CT density values282.283. and 284. and texture of the lung parenchyma. 285 In patients with pulmonary fibrosis, the density histogram is characteristically more peaked (kurtotic) and skewed to the left, than the normal distribution.283. and 284. The degree of kurtosis correlates moderately well with the severity of restrictive pulmonary impairment. 284 These histogram-based measures show changes over time indicating progression. 286 The proportion of lung with attenuation between −400 and −700 HU may correlate with severity of physiologic impairment. 287 An operator-independent technique that relies on fractal analysis of CT sections has been reported to have good accuracy for the quantification of fibrosing alveolitis. 288 Techniques for image analysis allow estimation of lung volumes, and simultaneous calculation of the relative amounts of soft tissue and air present in the lung. 282 Lung surface area may also be calculated. 289 Textural analysis of images may use multiple image features to differentiate between features of honeycombing and reticular abnormality,290. and 291. or to distinguish different types of lung disease such as emphysema, sarcoidosis, and IPF.292. and 293. Factors which limit the wide application of quantitative CT include the lack of spirometric gating, and substantial variation in reconstruction algorithms among manufacturers. 284
Many patients with IPF are cigarette smokers38 and thus emphysema is a common accompaniment. This combination has long been recognized as being responsible for spuriously preserved lung volumes on chest radiography (see Fig. 10.3A) and pulmonary function testing.30. and 31. On HRCT the morphologic differences between emphysema and fibrotic lung can usually be readily made (see Fig. 10.3B), although the distinction between the two processes in the mid-zones becomes difficult.
Prediction of reversibility and survival based on CT appearances
Many studies of CT in patients with IIP performed in the 1900s found that ground-glass abnormality was an important predictor of treatment responsiveness and/or survival.20.22.24.94. and 95. With our new understanding of the pathologic subsets of IIP, it seems clear that much of this effect was driven by the fact that ground-glass abnormality is the salient CT finding in NSIP and DIP, but is sparse or absent in patients with UIP. In patients with true UIP, ground-glass abnormality usually indicates fine lung fibrosis, and commonly progresses to honeycombing. In several studies of patients with UIP, the extent of honeycombing on CT,16. and 294. or the extent of fibrosis,286.295. and 296. correlates with poorer survival.
Given the prognostic importance of the histologic pattern, an important question is whether CT can perform as effectively as histology in predicting outcome. A study by Flaherty et al. 16 evaluated this issue in 168 patients presenting with the suspected IIP. Of these, 106 had histologic UIP, 33 had NSIP, and 22 had RB-ILD/DIP. The presence of honeycombing was strongly predictive of UIP, but its absence did not exclude UIP. The survival of patients with honeycombing on CT was significantly poorer than those without honeycombing. However, with regard to survival, the predictive value of honeycombing was less strong than that of histologic diagnosis. In a subsequent study, the same group found that, among patients with histologic UIP, those with a CT diagnosis of UIP (usually those with honeycombing) had significantly poorer survival than those with indeterminate CT (median survival 2.1 vs 5.8 years): the survival in those with UIP and indeterminate CT was in fact very similar to NSIP. 68
IDIOPATHIC UPPER LOBE FIBROSIS/FIBROELASTOSIS
Several case series have been published describing a condition characterized by progressive dyspnea associated with radiographic findings of progressive upper lobe predominant fibrosis, with marked apical pleural thickening and hilar retraction.297.298.299.300. and 301. This is sometimes associated with pneumothorax, and has been described as being very similar to the upper lobe fibrosis identified in ankylosing spondylitis. The syndrome has been described in three sisters. 302 The usual causes of upper lobe fibrosis such as tuberculosis, sarcoidosis, and hypersensitivity pneumonitis are excluded.
It has recently been hypothesized303 that these previously described cases of unexplained upper lobe fibrosis represented idiopathic pleuroparenchymal fibroelastosis, 304 a rare condition characterized histologically by marked thickening of the apical visceral pleura and prominent subpleural fibrosis, with a mixture of elastic tissue and dense collagen. The parenchyma distant from the pleura is spared. In a series of five patients with this condition, 303 with imaging available in four, the patients (including twin sisters) presented with clinical features suggestive of an IIP. The chest radiographs showed marked pleural thickening in all patients, with upper lung predominance and bilateral hilar retraction in two (Fig. 10.28). On CT, subpleural reticular abnormalities were found in two cases, centrilobular nodularity in two cases, with honeycombing and traction bronchiectasis each found in one case. In another two patients with pleuroparenchymal fibroelastosis, 305 the salient imaging features on chest radiograph and CT were apical subpleural fibrosis with volume loss. One of the individuals in this series experienced a spontaneous pneumothorax, followed by a postsurgical bronchopleural fistula which ultimately led to death. Individuals with this condition may be at risk for prolonged spontaneous or iatrogenic pneumothorax.
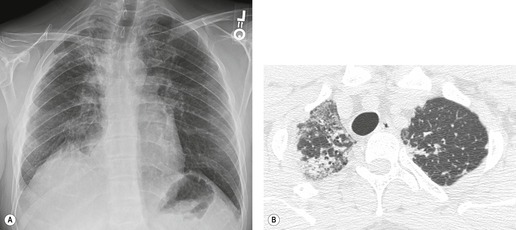
COLLAGEN VASCULAR DISEASE
Involvement of the respiratory system in the collagen vascular diseases (Box 10.20) is common and can result in considerable morbidity and mortality. The autoimmune-mediated inflammation and fibrosis that characterize these diseases in other organs can irreversibly disrupt the normal functioning of the lung. Many of these diseases are characterized by the presence of a specific type of autoantibody, which may greatly assist specific diagnosis (Table 10.3). The underlying mechanisms that account for the tissue changes encountered in this group of illnesses are unknown. As a group they can affect each portion of the lung, the pleura, alveoli, interstitium, vasculature, lymphatic tissue, and airways both large and small (Table 10.4). Commonly, more than one compartment is involved (Fig. 10.29). Most of the parenchymal manifestations of collagen vascular disease are similar to those found in IIPs4 (see Table 10.1, p 562), and can be classified using the same system. 306 While Table 10.4 attempts to show that certain patterns of pulmonary involvement are more characteristic of particular autoimmune diseases, it is prudent to consider all compartments of the lung at risk in every patient with collagen vascular disease. The lung disease associated with collagen vascular disease may precede the clinical presentation of the collagen disease by 5 years or more.
Box 10.20
• Each type of collagen vascular disease is associated with recognizable patterns of involvement
• May involve one or many compartments of the lung (e.g. pleura, vessels, parenchyma)
• Ancillary imaging clues to diagnosis are often visible
| Disease | Associated autoantibodies |
|---|---|
| Rheumatoid arthritis | Rheumatoid factor, anti-cyclic citrullinated peptide |
| Progressive systemic sclerosis (PSS) | Anticentromere antibody (limited PSS) |
| Anti-SCL-70 | |
| Mixed connective tissue disease | Anti-ribonuclear protein |
| Dermatomyositis/polymyositis | Anti-Jo-1 |
| Systemic lupus erythematosus | Anti-double-stranded (ds) DNA and anti-Sm |
| Antinuclear factor (less specific) | |
| Antiphospholipid antibodies | |
| Sjögren syndrome | Anti-SS-A (Ro) |
| Anti-SS-B (La) |
| ++++, frequent clinical association;+, entity is relatively uncommon; empty cells indicate that the entity is not described or rare. | ||||||
| RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; MCTD, mixed connective tissue disease; PSS, progressive systemic sclerosis; PM/DM, polymyositis/dermatomyositis; UIP, usual interstitial pneumonia; NSIP, nonspecific interstitial pneumonia; LIP, lymphoid interstitial pneumonia. | ||||||
| Pattern | RA | SLE | MCTD | PSS | PM/DM | Sjögren syndrome |
|---|---|---|---|---|---|---|
| UIP pattern | ++ | + | + | ++ | + | + |
| NSIP pattern | + | + | ++ | ++++ | ++ | + |
| Organizing pneumonia | ++ | + | + | + | ++ | |
| Diffuse alveolar damage | + | ++ | + | ++ | ||
| Lymphoproliferative disease (including LIP) | + | ++ | ||||
| Alveolar hemorrhage | + | +++ | + | + | + | |
| Upper lobe fibrosis | + | |||||
| Aspiration pneumonia | + | ++ | ++ | |||
| Pulmonary hypertension | + | + | + | ++ | + | |
| Bronchiectasis | ++ | ++ | ||||
| Obliterative bronchiolitis | ++ | + | ||||
| Nodules | ++ | |||||
| Pleural effusion | ++ | ++ | + | |||
| Muscle weakness/diaphragm dysfunction | + | + | ++ | |||
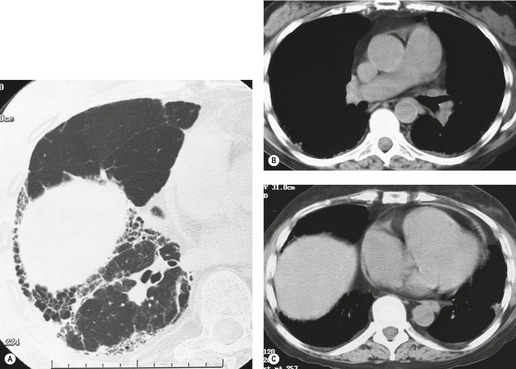
Careful evaluation of the chest radiograph and chest CT in patients with parenchymal abnormalities can yield some useful clues to the presence of collagen vascular disease. Joint abnormalities (shoulder or acromioclavicular) suggest RA. A dilated esophagus should suggest scleroderma or one of its variants (see Fig. 10.45). An enlarged pulmonary artery (out of proportion to extent of lung parenchymal abnormality) may be seen in many types of collagen vascular disease, particularly scleroderma (see Fig. 10.47). Soft tissue calcifications may be seen in dermatomyositis or scleroderma (see Fig. 10.51).
Rheumatoid arthritis
RA is a subacute or chronic inflammatory polyarthropathy of unknown cause that particularly affects peripheral joints. It is about three times more common in females than males and usually has an insidious onset, pursuing a variable course with relapses and remissions. The diagnosis of RA is based on the presence of clinical and laboratory features, including morning stiffness, arthritis of peripheral joints, positive rheumatoid factor, rheumatoid nodules, and erosions. 308 Anti-cyclic citrullinated peptide (anti-CCP) antibodies are highly specific markers for RA and useful for predicting RA development and progression. 309
In addition to an arthropathy, many patients have one or more extraarticular manifestations, such as subcutaneous nodules, ocular inflammation, pericarditis, splenomegaly, Felty syndrome, skin ulceration, and a variety of pleuropulmonary disorders. 307 The prevalence of these complications, particularly the pulmonary complications, depends on the diligence with which they are sought. Complications are more common in subjects who are seropositive, particularly with high rheumatoid factor titers, active erosive disease, and in patients with rheumatoid nodules. Smoking is also a risk factor for extraarticular abnormality, 310 and specifically for pulmonary fibrosis. 311 The 5-year mortality of patients with extraarticular manifestations of RA is about twice that of those without any extraarticular manifestation. 307 Definite and probable pleuropulmonary manifestations and associations of RA are listed in Box 10.21.
Box 10.21
• Pleural
– Pleuritis
– Effusion
– Pleural thickening
– Necrobiotic nodule
– Empyema
– Pneumothorax/bronchopleural fistula
• Interstitial pneumonia
– NSIP
– UIP
– Organizing pneumonia
– LIP
– Upper lobe fibrosis
• Nodule
– Necrobiotic nodules
– Caplan syndrome
– Lymphoid hyperplasia
• Pulmonary hypertension
• Airway disease
– Bronchitis
– Bronchiectasis
– Bronchiolitis: obliterative bronchiolitis; follicular bronchiolitis
• Drug-related disease
• Amyloidosis
• Lung cancer
CT findings in rheumatoid lung disease
There are a large number of papers describing the CT changes in rheumatoid lung disease.312.313.314.315.316.317.318. and 319. In the majority of studies of symptomatic and asymptomatic individuals, bronchial wall thickening and bronchiectasis are the most common findings. Variation in the prevalence of CT findings probably reflects differing CT techniques and diagnostic criteria. Abnormalities found on CT are often not associated with symptoms.
In an early study of 84 patients, the most common findings were bronchial wall thickening with bronchiectasis (30%), nodules (22%), nonseptal lines (18%), pleural opacity (16%), ground-glass opacity (14%), honeycombing (10%), and consolidation (6%). 312 In 36 patients with recent onset RA, 33% had interstitial abnormality on CT, more commonly in men. 314 In 126 patients with RA, bronchial dilation was found in 41%, ground-glass attenuation in 27%, parenchymal micronodules in 15%, reticulation in 12%, honeycombing in 9%, and consolidation in 4%. 320 Interstitial abnormalities were equally prevalent in those with early and longstanding RA, though bronchiolitis pattern was more prevalent in those with longstanding RA. In 75 unselected patients with RA, two-thirds of whom had no pulmonary symptoms, CT was abnormal in 49%, with interstitial abnormalities in 28%, and bronchiectasis in 19%. 321 Factors significantly associated with CT abnormalities were age older than 40 years, IgM rheumatoid factor, hypoxia at rest, and physiologic evidence of distal airway disease. 321 In a study of 34 patients with early RA (duration <1 year), CT showed expiratory air-trapping in 69%, bronchiectasis in 58%, and ground-glass opacity in 35%. 322 In 52 unselected patients, CT was abnormal in 67%, with reticulonodular abnormality in 63%, ground-glass attenuation in 20%, and bronchiectasis in 17%. 323 An interesting recent study324 evaluated CT findings in 14 asymptomatic first-degree relatives of individuals with known RA. Six of seven individuals who had positive RA-related antibodies had expiratory air-trapping, compared with none of seven antibody-negative subjects. This intriguing finding suggests that the airways or the lung may be involved very early in the evolution of RA.
Pleural disease
At postmortem examination, there are pleural changes in some 50% of cases of RA. 325 About 20% of patients with RA experience an episode of pleurisy. 326 However, clinical pleural effusion (Box 10.22) is much less common, and in a study of 516 patients with RA, only 17 (3.3%) had pleural effusions, with a higher prevalence in males (7.9%) than in females (1.6%). 327 Another large series found a 3.3% prevalence and an annual incidence of effusion of about 1% in patients with rheumatoid disease. 328 Unlike rheumatoid disease in general, but in common with other pulmonary manifestations of the disease, pleural effusion shows a striking preponderance of male patients.329. and 330. Patients are usually middle aged, with a mean age of about 50 years, with high titers of rheumatoid factor, longstanding active articular disease and rheumatoid nodules.326. and 329. The effusions in rheumatoid disease, unlike those seen in SLE, are commonly asymptomatic. 331
Box 10.22
• Often chronic
• Usually unilateral
• Low glucose level is characteristic
• Empyema may occur
Pleural effusions (Fig. 10.30) may be present at any phase of RA, 322 may predate clinical disease,329. and 330. or may even be present without arthritis. 332 In about one-third of patients there will be other rheumatoid-related abnormalities on the chest radiograph such as interstitial pneumonia and fibrosis or parenchymal nodules. 329 Effusions are most commonly small to moderate in size329 though they can be large.333. and 334. About one-fifth occur bilaterally. 329 Once formed, the effusions behave in a variable fashion, and although some resolve within weeks, more characteristically they persist for months and indeed sometimes for several years. 329 They often become loculated. Effusions may recur on the same or opposite sides of the chest. 335 Effusions may be associated with pneumothorax.336. and 337. Once the effusion has cleared, it commonly leaves residual pleural thickening,328. and 330. fibrothorax, and round atelectasis, 325 which may warrant decortication.327. and 338. Probably because of the high cellular content, rheumatoid pleural effusions may be intensely hypermetabolic on FDG-PET scan. 339
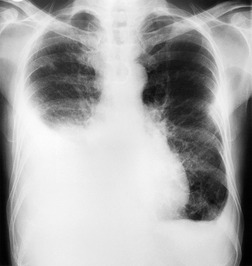
The diagnosis may be strongly suspected or established by the findings in the pleural fluid,325.330. and 340. and sometimes by pleural biopsy, which, although frequently nonspecific, may show rheumatoid nodules. The most characteristic features of the fluid are high cellular content, low sugar, low pH, and raised lactic dehydrogenase level. Rheumatoid factor is often present but is unfortunately a nonspecific finding. 340
Interstitial pneumonia and fibrosis
Lung fibrosis was first associated with RA by Ellman and Ball in 1948. 341 In a cohort of 609 patients with RA, the 30-year cumulative incidence of lung fibrosis was 6.8%. 310 The two commonest histologic patterns of lung fibrosis in RA are UIP (see p 564) and NSIP342 (see p 572). 343 Organizing pneumonia may also be seen.157.200.344.345. and 346. A few cases of DIP have been described.346. and 347. Lymphoid aggregates with germinal centers may be seen, 344 and, though nonspecific, are suggestive of rheumatoid lung. Increased numbers of T cells and B cells are present in the pulmonary lesions.348. and 349. When lymphoid aggregates are seen adjacent to bronchioles, follicular or lymphocytic bronchiolitis is diagnosed.
Since most patients with lung fibrosis related to RA do not undergo lung biopsy, it is difficult to determine the precise prevalence of the histologic subtypes of interstitial pneumonia in these patients. Katzenstein et al. 129 suggested that UIP was more common than NSIP in patients with rheumatoid disease. A study of 63 patients with rheumatoid lung, 17 of whom had undergone lung biopsy, identified histologic NSIP in 10 cases, UIP in two, organizing pneumonia in two, diffuse alveolar damage in one, DIP in one, and lymphocytic bronchiolitis in one patient. 343 However, among patients who did not undergo biopsy, a CT pattern of UIP (n = 22) was more common than NSIP (n = 11) or organizing pneumonia (n = 3), probably reflecting a lower threshold for performing lung biopsy in patients with an imaging pattern of NSIP as compared with UIP. A study of 16 patients with RA evaluated by surgical lung biopsy identified a histologic UIP pattern in seven, NSIP in seven, and mixed UIP and NSIP in two. 342
Rheumatoid lung fibrosis is substantially more common in men than in women,314. and 347. with a mean age of onset of about 50 years. 329 A higher proportion of male cigarette smokers may account for some of the increased risk among men. Most cases occur after the onset of the arthritis, though some 15–30% occur before or coincident with the initial joint manifestations.347. and 350. In general, whichever develops first, the interval between the onset of arthritis and lung disease is not more than 5 years. 329 Respiratory symptoms can be absent despite imaging changes351 but, if present, consist chiefly of exertional dyspnea and cough.
The prognosis of interstitial pneumonia in rheumatoid disease varies from series to series. Some studies show that the survival is as poor as that seen in idiopathic forms of lung fibrosis,347. and 352. while other authors have suggested that it is less severe.331. and 353. A recent study indicated that the prognosis was similar to IIP, when adjusted for age. 354 The difference between these series is probably related to the clinical and histologic heterogeneity of rheumatoid lung fibrosis. In one series, the mean duration of lung disease to death was 5 years, with a range of 1–17 years. 347 A subset of patients has a fulminant course, dying within months of the initial diagnosis, presumably because of acute exacerbation or AIP.114.355. and 356. Treatment with steroids or immunosuppressants helps improve physiologic and imaging findings of disease in a proportion of the patients, at least in the short term.347. and 356.
The chest radiographic abnormalities in rheumatoid lung fibrosis are indistinguishable from cases of IIP (Fig. 10.30). Pleural effusion or thickening may coexist with interstitial changes in about 5–15% of cases.329. and 347. Pulmonary nodules may also be seen in association with lung fibrosis. 357 CT findings in interstitial pneumonia associated with RA (Figs 10.31 and 10.32) are similar to those of the idiopathic variety.313.358. and 359. However, associated nodules, mosaic attenuation, pulmonary arterial enlargement, and pleural abnormality may provide a clue to the underlying diagnosis (see Fig. 10.29). In a study of 63 patients with rheumatoid lung disease, 343 26 had a CT pattern suggestive of UIP, 19 had a pattern of NSIP, 11 had a bronchiolitis pattern, and five had an organizing pneumonia pattern (Fig. 10.32). These CT patterns were in agreement with the histology in 13 of the 17 who underwent biopsy. Two patients who were classified as UIP by CT received a pathologic diagnosis of NSIP, one patient who was classified as NSIP received a pathologic diagnosis of DIP, and one patient with a CT diagnosis of LIP was classified pathologically as NSIP. Organizing pneumonia occurring in patients with RA is characterized on chest radiograph and on chest CT by patchy parenchymal opacities that may be migratory.157. and 345. Drug toxicity and infection should always be considered in the differential diagnosis of this appearance.
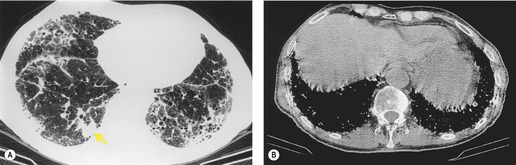
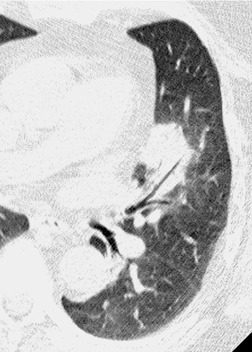
Necrobiotic nodules
Necrobiotic nodules occurring in the lung are pathologically identical to subcutaneous rheumatoid nodules347 and consist of a necrotic center bounded by palisading histiocytes (epithelioid cells) and surrounded by plasma cells and lymphocytes. Peripherally there may be a moderate, non-necrotizing vasculitis. 362 Intrapulmonary nodules are rare findings on the chest radiographs of patients with rheumatoid disease; two large series of 955 patients contained no examples;363. and 364. and another contained only two cases in 516 patients. 329 Nodules, like pleural effusions and IPF, are more common in men than women, with about a twofold excess. Mean patient age at presentation is 51 years (range 24–64 years). Nodules usually occur in patients with established disease but may occur with or before the onset of arthritis.347.365. and 366. Some patients with histologically proven nodules remain seronegative and never develop arthritis.366. and 367.
Nodules are usually asymptomatic. 368 Occasionally, symptoms result from superadded infection, 369 bronchopleural fistula formation, or pneumothorax. 370 Subcutaneous nodules are found in 80% of those with pulmonary necrobiotic nodules.329.335. and 368. Serum rheumatoid factor is found in nearly 90% of patients with nodules.335. and 368.
The nodules (Fig. 10.33) are usually discrete on imaging, rounded or lobulated, and often subpleural. 358 They may be single or, in about three-quarters of patients, they are multiple, 347 sometimes coalescing, 358 and show some mid/upper zone predilection. 366 They range in size from a few millimeters347 to 7 cm. Occasionally, when the nodules are small and widely disseminated, a miliary pattern is produced. 335 In about 50% of cases, the nodules cavitate (Fig. 10.34),335. and 347. producing ring opacities often with relatively smooth thick walls.335.371. and 372. Cavitation is more easily appreciated on CT. 358 Occasionally, nodules calcify.360. and 371. Subpleural nodules may erode through the pleura, and even into a rib373 and cause a bronchopleural fistula and hydropneumothorax.335. and 374. Nodular lesions may increase in size and number, resolve completely, wax and wane,375. and 376. or remain stable for many years. 347 Unless nodules are indolent or resolve, on imaging they are indistinguishable from pulmonary neoplasms and need either careful follow-up for increasing size, or histologic confirmation. 377 Cytologic evaluation of fine needle aspiration can be diagnostic. 378 Small rheumatoid nodules have also been described in the trachea379 and pleura.
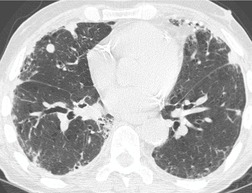
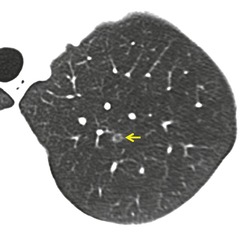
CT is clearly more sensitive for lung nodules than chest radiographs. The prevalence of nodules has been recorded in various CT studies as 0%, 314 6%, 313 10%, 315 22%, 312 28%, 380 and 37%. 351 The larger figures contain a high proportion of micronodules. Remy-Jardin et al. 312 characterized nodules into three categories:
• Parenchymal micronodules (7%) connected to branching bronchovascular structures, presumably representing follicular or constrictive bronchiolitis
• Subpleural micronodules (rounded, hemispheric, or triangular and less than 3 mm diameter) which sometimes coalesced into plaquelike lesions, which might represent foci of lymphoid hyperplasia, pleural thickening or necrobiotic nodules.
Airways disease
The increasing use of CT in patients with RA has made it plain that airways disease is one of the most common patterns of abnormality seen in these patients (Box 10.23). 382 Both bronchitis (airway wall thickening) and bronchiectasis are more frequent in patients with rheumatoid disease than in matched controls. 340 Bronchiectasis may precede or follow the onset of rheumatoid disease.383. and 384. In CT-based studies, an overall frequency of bronchiectasis of about 20% is recorded, with a range of 6–41%.312.314.315.318.320. and 380. Bronchiectasis may be unassociated with respiratory symptoms. 318 Bronchiectasis is usually cylindric in type, and is commonly, though not always, associated with CT and physiologic evidence of small airways disease (Fig. 10.35). 385
Box 10.23
• Bronchitis
• Bronchiectasis
• Obliterative bronchiolitis
• Follicular bronchiolitis
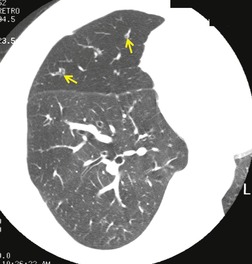
There is a recognized association between rheumatoid disease and obliterative bronchiolitis (constrictive bronchiolitis)199.360.386.387. and 388. in which bronchioles are destroyed and replaced by scar tissue. Although penicillamine or gold has been implicated in the pathogenesis of obliterative bronchiolitis, it also occurs in patients taking neither drug. 340 Clinically there is progressive dyspnea and evidence of irreversible airflow obstruction. The chest radiograph is either normal or shows hyperinflation, with or without areas of oligemia. The characteristic CT finding is mosaic perfusion (Fig. 10.35) with air-trapping,389.390.391. and 392. often associated with evidence of mild bronchiectasis.
Follicular bronchiolitis is a second type of small airway disorder recognized in rheumatoid lung disease.319.344.346.393. and 394. It is characterized by lymphoid aggregates, with or without germinal centers, lying in the walls of bronchioles and possibly compressing their lumens. 395 Follicular bronchiolitis probably produces a reticular or reticulonodular pattern on the chest radiograph. 344 The major CT finding is centrilobular nodules, often associated with peribronchial nodules, and with areas of ground-glass abnormality.319. and 395. HRCT may sometimes be normal. 393 Should lymphoid hyperplasia in rheumatoid lung disease be more concentrated along septa and subpleural areas than in airways, the term lymphoid hyperplasia is used. 346 Radiographically it gives rise to reticular or reticulonodular opacities344 but its HRCT features are not described; Remy-Jardin et al. 312 suggested that it may account for subpleural micronodulation, a common finding in rheumatoid lung disease.
Diffuse panbronchiolitis has been described in patients with RA, 396 but this could well be a chance association. A handful of patients with rheumatoid disease have had biopsy-proved bronchocentric granulomatosis (see Chapter 11, p 674).397.398. and 399. On imaging, there were bilateral nodules and focal consolidations ranging in size from 2 cm to 15 cm, some of which were cavitated.
Caplan syndrome
The original description of Caplan syndrome was of multiple, large (0.5–5.0 cm in diameter) rounded nodules seen on the chest radiographs of coal miners with RA. 400 Radiographic evidence of simple coal miner’s pneumoconiosis was often absent. The syndrome may occur with exposure to inorganic agents other than silica or coal. 401 Caplan nodules are pathologically similar to necrobiotic rheumatoid nodules. 402
Although Caplan syndrome was quite common in the UK, 403 the prevalence in the USA has always been low, 404 and only a single English-language case report of this entity appears to have been published since 1965. 405
The classic imaging finding in Caplan syndrome is unilateral or bilateral pulmonary nodules (Fig. 10.36), usually 1–2 cm in diameter, but ranging from 0.5 cm to 5 cm. 400 Imaging changes of coal worker’s pneumoconiosis may or may not be present. 400 Nodules are typically situated at the junction of the outer and middle thirds of the lung and tend to appear in crops of lesions having a similar size and rate of growth. Nodules tend to develop rapidly and grow over a period of months. They may heal with scarring, remain stable or grow slowly for several years. Calcification or cavitation may be seen on chest radiograph403 or CT. 405 Smaller Caplan nodules may be indistinguishable from silicotic nodules. 405
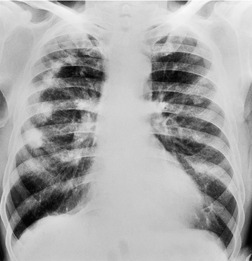 |
| Fig. 10.36 (Courtesy of Dr. P M Hacking, Newcastle upon Tyne, UK.) |
Pulmonary vasculopathy
Systemic vasculitis in rheumatoid disease is usually of the small vessel type and affects chiefly the skin. There is some evidence that the prevalence of vasculitis in RA has increased with increasing use of new treatments targeted at tumor necrosis factor. Pulmonary involvement is relatively uncommon.406.407.408. and 409. Prognosis is poor and most patients have died within a year of diagnosis. 409 Rheumatoid pulmonary vasculitis must be distinguished from Wegener granulomatosis, which may also occur in patients with RA. 410
Pulmonary hypertension commonly occurs in patients with RA, but is usually mild. 411 It increases in prevalence with increasing duration of RA. 412 It may be out of proportion to the visible extent of lung disease, suggesting the presence of an independent pulmonary vasculopathy. It should be suggested when the pulmonary arteries appear enlarged on chest radiograph or chest CT (see Fig. 10.29). Pulmonary hemorrhage is very uncommon in patients with RA.
Other associations
Most studies show that RA is associated with an increased incidence of lymphoma, and of lung cancer (see Fig. 10.31). 413
Many of the available treatments for RA, including gold, methotrexate and D-penicillamine have been implicated in the development of infiltrative lung disease (Table 10.5). 414 On chest CT scans, gold-induced lung disease was characterized in 12 of 20 reported cases by alveolar opacities extending along bronchovascular bundles. 415 Low-dose methotrexate may be associated with subacute hypersensitivity pneumonitis in 2–5% of cases (Fig. 10.37).416. and 417. Preexisting radiographic evidence of ILD probably predisposes to the development of methotrexate pneumonitis in patients with RA. 418 Treatment with D-penicillamine is associated with the development of constrictive bronchiolitis. Nonsteroidal anti-inflammatory drugs may be associated with hypersensitivity reactions (Fig. 10.38),419. and 420. while unintentional therapeutic overdoses of salicylate may result in pulmonary edema. Leflunomide, a pyrimidine synthesis inhibitor, may cause a wide array of pulmonary complications, including DAD, acute eosinophilic pneumonia, and COP. 421
| Aspirin (excessive use) | Lung edema |
| Nonsteroidal anti-inflammatory drugs | OP pattern |
| Gold | OP pattern |
| Methotrexate | NSIP pattern |
| Penicillamine | Bronchiolitis |
| Cytotoxic drugs | Opportunistic infection |
| Antitumor necrosis factor agents | Mycobacterial or fungal infection, rarely AIP |
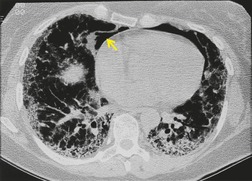
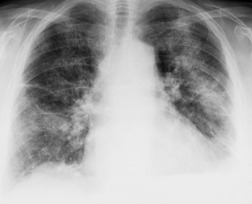
The new generation of biologic agents used to treat RA have resulted in a new array of potential pulmonary side effects. The most important of these is impaired immunity related to use of antitumor necrosis factor (TNF)α antibodies (etanercept, infliximab, and adalimumab), which results in a substantially increased incidence of tuberculosis (sometimes disseminated or extraarticular) and nontuberculous mycobacterial infection. 422 Fungal and pneumocystis infection may also occur. Screening chest radiographs are usually obtained when anti-TNFα antibody treatment is planned. Mycobacterial or fungal infection should be strongly suspected when new parenchymal abnormalities are identified in these patients.
Systemic lupus erythematosus
SLE is a multisystem collagen vascular disorder that particularly affects vessels, serosa, joints, kidneys, central nervous system (CNS), skin, and blood elements. Its manifestations are extremely variable as regards both organs involved and severity. A characteristic feature is autoantibody production against a wide variety of cellular constituents including nuclear material, particularly deoxyribonucleic acid (DNA). In a series of 1000 patients the five commonest autoantibodies were antinuclear antibody (ANA) in 96% of cases, anti-double-stranded DNA (anti-dsDNA) (78%), anti-Ro (25%), anti-La (19%), and anti-Sm (10%). 423 Two autoantibodies – anti-dsDNA and anti-Sm are considered diagnostic, with high specificity but only moderate sensitivity. Because the manifestations of the disease are so variable, diagnostic criteria have been established (Box 10.24). Should any four of the criteria be present simultaneously or serially during a period of observation, then SLE can be diagnosed. 424
Box 10.24
• Malar rash
• Discoid lupus erythematosus
• Photosensitivity
• Ulceration of mouth and oropharynx
• Arthropathy (nonerosive, nondeforming)
• Serositis
• Renal disorder (proteinuria or cellular casts)
• Neurologic disorder (epilepsy or psychosis)
• Hematologic disorder (hemolytic anemia, leukopenia, lymphopenia, thrombocytopenia)
• Autoantibodies
• Antinuclear factor (less specific)
• Anti-Sm
• Anti-dsDNA
• Antiphospholipid antibody
The overall prevalence of SLE is about 20–50 cases per 100 000 population and it is 10 times as common in women as in men425 with an increased prevalence among relatives and black people. The illness characteristically shows relapses and remissions, with a tendency to progression and eventual multiorgan dysfunction. 426 It presents typically in women of childbearing age, with a wide variety of manifestations, particularly those of joint, skin (malar rash, photosensitivity, livedo reticularis), and systemic disorders. The prevalence of the various clinical manifestations in the four series426 was: arthropathy 85%, skin lesions 80%, nephritis 53%, pleurisy 52%, neuropsychiatric disorders 44%, lymphadenopathy 43%, pericarditis 39%, and mucosal ulceration 15%. About 10% of patients present over the age of 50 years, with features that resemble SjS or polymyalgia rheumatica. 423 These patients are less likely to present with the typical malar rash or arthritis. There is usually an anemia, leukopenia, and thrombocytopenia – a combination that is unusual in other inflammatory conditions. 427 The reported prognosis has greatly improved over the years, in part due to better therapy but also because less severe or subclinical cases have been included in the various studies. In a recent series, the 5-year survival was 98%, and 10-year survival was 89%. 428
Thoracic involvement is common in SLE, and the lungs, pleura, heart, diaphragm, and intercostal muscles may all be affected. 429 Thoracic manifestations (Table 10.6) can be divided into primary changes or secondary complications, but this distinction is by no means always clear-cut. Cardiac involvement may include pericarditis, endocarditis, myocarditis, coronary artery disease, and vasculitis. 430 Pleuropulmonary involvement in SLE may be asymptomatic or minimally symptomatic. In a multiethnic cohort of 626 patients with lupus, 431 46 (7.3%) patients had evidence of lung injury after a mean disease duration of 5.3 years: 25 had pulmonary fibrosis, 12 pulmonary hypertension, eight pleural fibrosis, four pulmonary infarction and four shrinking lung syndrome. Seven patients had more than one type of lung damage. However, the degree of pulmonary impairment in these patients is not clear.
| Category of abnormality | Clinical entity |
|---|---|
| Serositis | Dry pleuritis |
| Pleural effusion | |
| Pericarditis | |
| Acute parenchymal abnormality | Diffuse alveolar damage |
| Acute lupus pneumonitis | |
| Diffuse pulmonary hemorrhage | |
| Subacute or chronic interstitial pneumonia | Nonspecific interstitial pneumonia |
| Organizing pneumonia | |
| Usual interstitial pneumonia | |
| Pulmonary vascular | Pulmonary arterial hypertension |
| Vasculitis/capillaritis | |
| Antiphospholipid syndrome with pulmonary embolism | |
| Pulmonary venoocclusive disease | |
| Miscellaneous | Diaphragmatic dysfunction |
| Opportunistic infection | |
| Atelectasis |
Pleuritis/pleural effusion
Pleuritis is found in 40–60% of patients with SLE and it is the commonest pleuropulmonary manifestation.432.433. and 434. It may be a presenting feature, 435 but occurs more commonly during an exacerbation of established disease. 331
The pleuritis is dry 50% of the time, 436 but in the other 50% it is accompanied by a pleural effusion and sometimes also by a pericardial effusion (Fig. 10.39).429. and 437. The pleural effusions are usually small or moderate in size. 437 Unilateral and bilateral effusions are equally frequent.435. and 437. Sometimes the pleural effusions resolve spontaneously, but many require treatment with steroids or immunosuppressive drugs. Clearing may be complete or incomplete, leaving minor pleural thickening.435. and 438. It is important to exclude other causes of pleural effusion in SLE, including the nephrotic syndrome, cardiac and renal failure, pulmonary embolism, and infective pneumonia.325. and 439. The fact that primary pleural effusions in SLE are almost always painful436. and 437. can be a helpful differentiating feature.
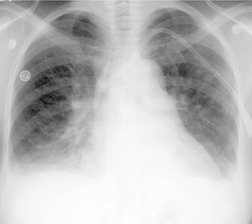
Acute lupus pneumonitis
Acute lupus pneumonitis is a poorly defined entity, characterized by a variable degree of respiratory impairment accompanied by focal or diffuse pulmonary consolidation, occurring in patients with lupus. 441 It is now believed that most cases previously identified as lupus pneumonitis probably represented AIP with or without pulmonary hemorrhage. 442 The alveolar damage is most likely mediated by immune complex deposition. 441 The clinical, histologic, and imaging features overlap substantially with those of pulmonary hemorrhage (Box 10.25; Fig. 10.40).
Box 10.25
• Infection
• Hemorrhage
• Lupus pneumonitis/DAD
• Pulmonary infarction
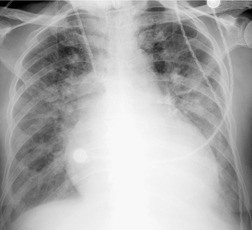
Pulmonary hemorrhage
Pulmonary hemorrhage is common in the lungs at postmortem examination in patients with SLE, 443 but may not be recognized clinically. Lung hemorrhage ranges from a mild, chronic, subclinical process to one that is acute and life-threatening. 441 In a large series of more than 400 patients with SLE at the National Institutes of Health (NIH), the prevalence of acute hemorrhage was only about 1.5%, 444 occurring typically in young (mean age ∼30 years) women. In 10–20% of patients with hemorrhage it is the first manifestation of SLE.445. and 446. Acute pulmonary hemorrhage is an important diagnosis to make since urgent treatment is needed. The pathologic findings are diffuse alveolar hemorrhage with acute necrotizing capillaritis.362. and 445. A minority of cases may be associated with DAD. 445 Bland pulmonary hemorrhage appears to be rare.
Clinically, in acute hemorrhage, there is rapid onset of severe dyspnea, fever, and hemoptysis. However, hemoptysis is absent in 33–50% of cases on admission. 447 Pulmonary hemorrhage is typically accompanied by signs of active disease elsewhere; fever, arthropathy, and particularly nephritis (14 out of 15 patients in one series).445. and 448. Helpful clinical pointers include hemoptysis and a 3–4 g drop in blood hemoglobin level. 447 Confirmation can be obtained by bronchoalveolar lavage.
The radiographic appearances are indistinguishable from other forms of pulmonary hemorrhage and consist of airspace opacity that is usually bilateral and diffuse (Fig. 10.41).448. and 449. The opacities usually clear within days. 450 CT shows patchy or diffuse parenchymal consolidation and/or ground-glass abnormality. Acute pulmonary hemorrhage must be differentiated from infection, and from lung edema due to renal or cardiac failure. In one series of 13 patients with 14 episodes of pulmonary hemorrhage, infection was identified in eight, with organisms including Pseudomonas and Aspergillus. 451
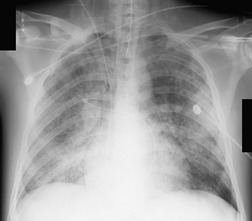
Lung fibrosis
Fibrotic ILD (Fig. 10.42) is less common in SLE than in the other collagen vascular diseases. 372 However, in four autopsy series, chronic interstitial abnormalities were present in just over one-third of cases. 443 In a selected clinical series of 30 patients with pleuropulmonary SLE, there was a 13% frequency of interstitial pulmonary fibrosis judged on imaging (with biopsy confirmation in two), 452 and in an unselected outpatient series assessed radiographically there was a 3% prevalence of interstitial pulmonary fibrosis. 453 A number of patients with interstitial pulmonary fibrosis have had preceding acute pneumonitis, 454 and the interstitial changes presumably represent the organizing phase of diffuse alveolar damage. The histology of other forms of interstitial pulmonary fibrosis is that of interstitial pneumonia, which is likely to be a mixture of UIP and NSIP, the latter probably being more frequent. 362 In general, interstitial disease in SLE pursues an indolent course, but there are exceptions and it may cause death.
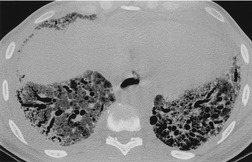
There is limited information on CT changes in SLE. In a study of patients with SLE who did not have previously diagnosed lung disease, Bankier et al. 455 found that 17 of 45 patients with a normal chest radiograph had an abnormal HRCT. Interlobular and intralobular lines were the commonest type of abnormality, being seen in 15 patients. Air space nodules, architectural distortion, bronchial wall thickening, bronchial dilatation, pleural irregularity, ground-glass attenuation and airspace consolidation were less common. Middle and lower lung zone involvement was predominant. The extent of abnormality on CT correlated with the length of the clinical history, and with impaired pulmonary function (decreased FEV1/FVC ratio and DLco). Fenlon and co-workers456 studied 34 consecutive patients with SLE, 56% of whom had never smoked and 23% of whom had respiratory symptoms. The commonest signs were thickened interlobar septa (44%), parenchymal bands (44%) (Fig. 10.43B), subpleural bands (21%), and pleural tags and thickening (15%). Only 6% had ground-glass opacity, consolidation, or honeycombing (Fig. 10.42). These interstitial changes were usually mild in degree, less commonly moderate and rarely severe. Overall, 33% of patients were judged to have mild or moderate ILD. In 21% of patients there was bronchiectasis and bronchial wall thickening. In contrast to the study by Bankier et al., 455 Fenlon et al. 456 did not confirm a relationship between the presence or extent of lung abnormality and pulmonary physiology or duration of disease. Because the extent of disease on CT in patients with SLE is usually relatively minor, the clinical value of CT in asymptomatic patients is questionable.
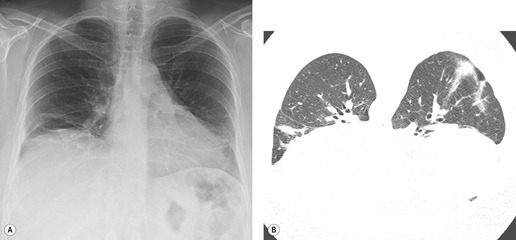
Diaphragm dysfunction
Diaphragm dysfunction in SLE may be manifest as elevation of one or both hemidiaphragms (Fig. 10.43) and is a cause of dyspnea. Bilateral elevation is a common finding and, in some series, has been the most common imaging pleuropulmonary abnormality in SLE, being seen in as many as 18% of patients. 428 As the diaphragm rises, the lungs lose volume, hence the term ‘shrinking lungs’, 457 a finding first noted in 1954. 432 The loss of lung volume was initially ascribed to reduction in lung compliance, but more recent consensus suggests it is due to diaphragmatic weakness458 presumably caused by a myopathy,459. and 460. for which there is some pathological evidence. 461 Pleuritic pain with splinting may be a contributory factor.459. and 462. Most patients stabilize or improve over time. 462 CT is helpful in excluding lung fibrosis as a cause of decreased volumes, and usually shows basal atelectasis adjacent to the elevated hemidiaphragms (Fig. 10.43). On fluoroscopy, diaphragmatic motion may be sluggish, or paradoxical upward movement of the diaphragm may be seen with inspiration or sniffing.
Pulmonary arterial hypertension/vasculitis
A variety of changes are described pathologically in the pulmonary vessels.362. and 463. Chronic lesions consist of intimal thickening, subintimal fibrosis, medial hypertrophy, and periadventitial fibrosis. Plexogenic lesions similar to those of primary pulmonary hypertension, and organizing thrombi may also be seen. 464 Marked pulmonary arterial hypertension is unusual. 429 Mild pulmonary artery hypertension is quite common465 and in one study with echocardiographic assessment on two occasions, separated by 5 years, there was respectively a 14% and 43% frequency of pulmonary artery hypertension with relatively modest mean pressures of 23 mmHg and 28 mmHg. 466 Pulmonary artery hypertension is associated with digital ischemia, livedo, 467 and, in 75% of patients, Raynaud phenomenon, 468 findings that are consistent with a vasospastic mechanism. 469 Some cases of pulmonary arterial hypertension have been associated with the presence of lupus anticoagulant, discussed below. 468 In these cases, it is very important to exclude central pulmonary thromboembolic disease.
Antiphospholipid syndrome
Up to two-thirds of patients with SLE test positive for antiphospholipid antibodies, 442 and in a literature review of more than 1000 patients, 34% had lupus anticoagulant and 44% anticardiolipin. 470 The presence of antiphospholipid antibodies is an important risk factor for thrombosis (venous and arterial), neurologic disease, thrombocytopenia, and fetal loss. Subjects with SLE and antiphospholipid antibodies have a sixfold increased risk of venous thromboembolism, compared with those with SLE but no antibodies. 442 The risk is greater with the lupus anticoagulant than with the anticardiolipin antibody. A review of the thoracic imaging features of 88 patients with antiphospholipid antibodies471 found that pulmonary embolism was the primary intrathoracic manifestation, being seen in nine patients, usually associated with thrombosis of lower extremity veins. Thrombosis of the superior vena cava or its tributaries471 may also occur.
The antiphospholipid syndrome, which may occur without other manifestations of SLE, and may occur in other collagen vascular diseases, is defined by the occurrence of venous or arterial thrombus, and/or fetal loss, associated with the presence of one of the antiphospholipid antibodies. 442 The antiphospholipid antibody syndrome may cause thromboembolic pulmonary hypertension.472. and 473. Pulmonary arterial aneurysms, presumably due to vasculitis, have also been described in this syndrome. 474 Other manifestations may include nonthrombotic pulmonary hypertension, 475 lung fibrosis, 476 acute interstitial pneumonia, and cardiac valvular lesions. 442 Diffuse alveolar hemorrhage (Fig. 10.44) 477 is quite common in our experience. Antiphospholipid syndrome should be suspected in patients with pulmonary embolism or pulmonary hypertension in whom there is no evident predisposing factor, and particularly in those with thrombosis at unusual sites such as central veins or dural sinuses. 478 Magnetic resonance angiography may be helpful in visualizing the entire vascular system to document the extent of venous and arterial thrombi. 479 The catastrophic antiphospholipid syndrome consists of multiorgan microvascular occlusion, with a mortality rate of about 50%. 480
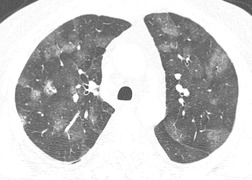
Miscellaneous manifestations
Organizing pneumonia has been recorded in a handful of patients.200. and 481. Acute reversible hypoxemia is thought to be caused by polymorph aggregation in the pulmonary circulation following activation by complement split products. The chest radiograph is normal and the condition resolves with steroid therapy. 482Pulmonary venoocclusive disease has been recorded. 483
Secondary manifestations
Atelectasis
Elevation of the diaphragm with resultant lower zone underinflation may be associated with basal line or band shadows that are usually horizontal, several millimeters wide, and up to 5 cm long. These most likely represent discoid or plate atelectasis, and may be related to diaphragmatic weakness, splinting from chest pain, or pulmonary infarcts. 429
Pneumonia
Infection should be the first consideration in all SLE patients, particularly those receiving an immunomodulatory agent, presenting with new/worsening respiratory symptoms or imaging abnormalities. 442 Pneumonia is probably the single most common pulmonary abnormality in SLE, 331 occurring in about 50% of patients.439.443. and 484. Most cases are simple bacterial pneumonias, 483 but some are opportunistic infections. 439 These latter are an increasingly important cause of death.441. and 485. The responsible organisms that have been recorded include Candida, Cytomegalovirus, Legionella, Pneumocystis, Cryptococcus, Aspergillus, and Nocardia.429.436.439.443.444. and 485. Cavitary lesions, which are uncommon in SLE, are most often the result of infections. 486
Pericarditis, myocarditis, and renal disease
Twenty to thirty per cent of all SLE patients develop pericarditis at some time. 430 A reliable figure for myocarditis is not available but is probably in the order of 8%. 433 Myocardial abnormalities were found in most patients with active SLE in a small series. 487 Myocarditis, however, rarely gives rise to cardiac failure. There is an 18–74% prevalence of valve disease (Libman–Sachs endocarditis, with valvular thickening, stenosis/regurgitation, and verrucose endocarditis), the frequency depending on the mode of diagnosis. 488 There is also increased risk of infective endocarditis. Renal failure and the nephrotic syndrome may cause pulmonary edema and pleural effusion. The pleural effusions, unlike those due to lupus pleuritis, are pain-free.
Progressive systemic sclerosis
Systemic sclerosis is a generalized connective tissue disorder characterized by: tightening, induration, and thickening of the skin (scleroderma); Raynaud phenomenon and other vascular abnormalities; musculoskeletal manifestations; and visceral involvement especially of the gastrointestinal tract, lungs, heart, and kidneys.489.490.491. and 492. The lung is the fourth most commonly affected structure after skin, vessel, and esophageal involvement. 493 In general, the extent of extrapulmonary involvement does not correlate with the severity of pulmonary fibrosis. 494 The diagnosis can be made with a high degree of certainty if the single major criterion of proximal scleroderma is present (proximal to metacarpophalangeal joints) or if there are two or more minor criteria (sclerodactyly, pitting scars or loss of substance of the finger tips, or bilateral basal pulmonary fibrosis). 495 Nailfold capillaroscopy can be very helpful in identifying the microcirculatory disturbance of scleroderma, and may be added to the diagnostic criteria. 496 Antinuclear antibodies are present in virtually every patient. The presence of anticentromere antibodies appears to be associated with the absence of ILD,497. and 498. while antitopoisomerase antibody is associated with the absence of pulmonary hypertension. 499 Pulmonary fibrosis is more common in African Americans. 497
The pathogenesis is complex and incompletely understood, the fundamental abnormality being persistent overproduction and tissue deposition of collagen and related macromolecules. 500 Factors that contribute to this are fibroblast proliferation, capillary endothelial damage causing increased vascular permeability, and an alveolitis.493.501.502. and 503. There are two types of histopathologic change in the lungs which are independent of each other: 504 interstitial fibrosis;36. and 37. and vascular changes in small vessels (intimal proliferation, medial hypertrophy, and myxomatous change). 362 In two series of patients with scleroderma who underwent lung biopsy, the prevalence of NSIP was about 75%, with UIP being found in 10–20%.36. and 37. At autopsy, the lungs are abnormal in at least 80% of cases. 505 The interstitial fibrosis may affect all lobes, but is particularly marked peripherally and in lower zones.362. and 506.
Scleroderma is classified clinically into five subsets: 507 in order of frequency, these are limited scleroderma (46%), diffuse scleroderma (33%), overlap syndrome (11%), undifferentiated connective tissue disease with scleroderma features (9%), and sine scleroderma (2%). Lung fibrosis is more common in diffuse scleroderma (56%) than in limited scleroderma (21%), while pulmonary hypertension is more common in diffuse scleroderma. 507 However, lung involvement occurs in all types of scleroderma, even in sine scleroderma, which is scleroderma without any cutaneous sclerosis. 508
Systemic sclerosis has a 3 : 1 female to male distribution and presents most commonly in the third to fifth decade of life although there is a wide range from childhood upwards. 495 The clinical syndrome of scleroderma may rarely occur in relationship to silicosis (Erasmus syndrome). 509 In children, a localized cutaneous form of the disease is the commonest manifestation, but pulmonary involvement does occur.510. and 511. The most frequent presentation in adults is with Raynaud phenomenon, which occurs in some 80–90% of cases and may precede skin changes by several years. Prognosis is very variable depending on the degree of visceral involvement, particularly of the heart, kidneys, and the lungs. 512 The 5-year survival is between 70% and 80%,491. and 513. and pulmonary disease has now replaced renal disease as the major cause of death.513.514. and 515.
Less than 1% of patients present initially with respiratory symptoms. 489 However, in established disease respiratory symptoms are common, with dyspnea in more than 60% and less commonly cough and pleuritic pain. 493 Dyspnea may be due to lung fibrosis and/or pulmonary arterial hypertension. 490 Pulmonary function tests are commonly (80–90%) abnormal in systemic sclerosis, but the defects are often mild. 489 The main abnormalities are reduction in carbon monoxide diffusion and a restrictive ventilatory defect, with hypoxemia and airflow obstruction in some.489. and 493. Restrictive lung disease is substantially more frequent in patients with the diffuse form of the disease, whereas pulmonary hypertension and impaired gas exchange are more prevalent in patients with limited scleroderma.499. and 516. Extent of disease on HRCT correlates with lung diffusing capacity (DLco). 517 About 50% of patients with systemic sclerosis have abnormal inflammatory findings on bronchoalveolar lavage (BAL). 493 Respiratory disease usually follows an indolently progressive course, and adversely affects survival. 514
The chest radiograph is abnormal in 10–80% of patients with established scleroderma514. and 518. (Box 10.26). As with other fibrosing lung diseases, the interstitial fibrosis of systemic sclerosis can be present despite a normal chest radiograph. 362 Occasionally, chest radiographic changes consistent with fibrosis antedate the onset of scleroderma, sometimes by many years. 519 The commonest radiographic abnormality is a widespread, symmetric, basally predominant reticulonodular pattern (Fig. 10.45)506. and 520. that typically starts as a very fine ground-glass or reticular pattern, often with traction bronchiectasis, and progresses to coarser reticulation, sometimes with honeycombing. 519 Loss of lung volume with elevation of the diaphragm often occurs and is caused in part by reduced compliance associated with fibrosis. Respiratory muscle abnormalities are common in systemic sclerosis and also play a part. 493 Pleural changes are infrequent and usually minor,493. and 506. even though at autopsy about a third of cases have pleural inflammation and adhesions. 505
Box 10.26
• Lung fibrosis (usually NSIP)
• Pulmonary hypertension
• Lung cancer
• Esophageal dilation
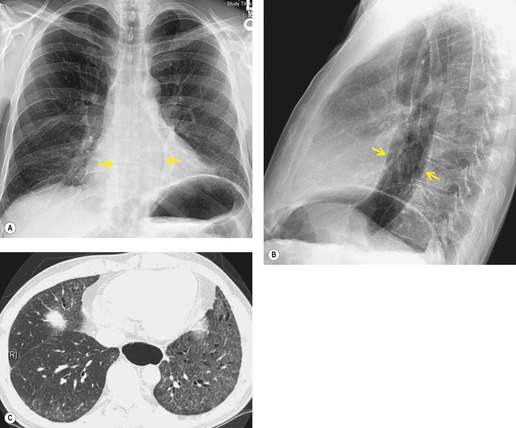
Involvement of the esophagus is present pathologically in three-quarters of patients with scleroderma. 514 The esophagus becomes functionally abnormal and fibrosed, ending up as a dilated, air-filled tube that may be detected on the frontal or lateral chest radiograph (Fig. 10.45).521.522. and 523. An air esophagogram is found in many other conditions that cause esophageal dysmotility, including other collagen vascular diseases and achalasia. 524 In contrast to patients with achalasia, the esophagus does not contain an air–fluid level, as the dilatation is not usually associated with obstruction. 525 Asymptomatic esophageal dilatation is more readily demonstrated (in up to 80% of patients) on CT. 526
CT findings in systemic sclerosis have been described in a number of papers.95.526.527.528.529.530.531.532.533.534. and 535. CT may be abnormal in the face of a normal chest radiograph.530. and 536. Changes, other than in advanced disease, tend to occur at the lung bases and posterolaterally.530. and 532. The main findings are confluent ground-glass opacification and fine reticular pattern, often posterior and subpleural, usually associated with traction bronchiectasis and bronchiolectasis (Figs 10.45 and 10.46).530. and 532. Honeycombing (usually minor) is seen in about 37% of symptomatic patients. 535 Not surprisingly, the CT findings in most cases of scleroderma are similar to those of NSIP, 537 since this is the histologic lesion in 75% or more of cases. Extent of lung disease identified on CT correlates moderately with physiologic impairment. 535 Extent of ground-glass abnormality and honeycombing correlates weakly with inflammatory change on bronchoalveolar lavage. 535 In a large treatment study, the extent of lung fibrosis identified on baseline CT was an important independent predictor of physiologic progression, and of response to treatment. 538 Quantitative CT methods are being developed to determine disease extent.539. and 540. There is an increased prevalence (32–60%) of enlarged reactive mediastinal lymph nodes in patients with systemic sclerosis, and this may be related to the extent of lung disease.526. and 528.
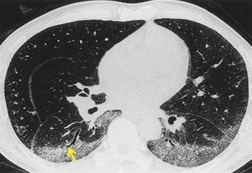
Pulmonary arterial hypertension, with pulmonary arterial enlargement, is common in systemic sclerosis,541. and 542. and occurs independently of lung fibrosis (Fig. 10.47) though both may occur together (Fig. 10.48).504. and 543. The finding of pulmonary arterial enlargement out of proportion to the severity of lung fibrosis is very suggestive of collagen vascular disease, particularly scleroderma. It occurs in one-third to one-half of patients particularly in anticentromere antibody ACA-positive patients with the CREST syndrome (limited cutaneous systemic sclerosis).504.544. and 545. Its presence may be signalled by an isolated reduction in diffusing capacity.544. and 545. Pulmonary arterial hypertension usually causes enlargement of the main and proximal pulmonary arteries on chest radiograph or CT, and may lead to cor pulmonale with cardiomegaly; however normal-sized pulmonary arteries do not exclude the diagnosis, and the presence of pericardial thickening or fluid in patients with scleroderma appears to be a stronger predictor of echocardiographic pulmonary hypertension (Box 10.26). 546
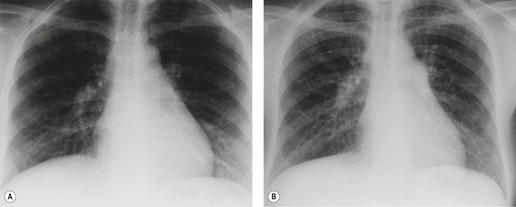
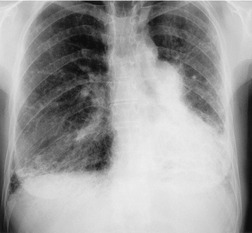 |
| Fig. 10.48 Systemic sclerosis with pulmonary fibrosis and pulmonary arterial hypertension. Unlike the patient illustrated in Fig. 10.47, here interstitial fibrosis and pulmonary arterial hypertension are occurring together, but the degree of pulmonary arterial enlargement is out of proportion to the degree of fibrosis. Mediastinal air to the left of the trachea and in the region of the left mainstem bronchus lies in a dilated dysmotile esophagus. |
There is an increased prevalence of malignancy in scleroderma, with relative risk of malignancy ranging from 1.8 to 6.5,514. and 547. although some studies have not shown an increased risk. 548 Lung cancer (Fig. 10.49) accounts for most of the increased risk, with a relative risk ranging from 5.9549 to 16.5550 and most, though not all, studies have indicated that the presence of lung fibrosis is a risk factor. 547 Smoking is the strongest risk factor. 551 The relative risk is probably about the same as is seen in patients with IPF (cryptogenic fibrosing alveolitis).413. and 493. All types of lung cancer may be present, but a slightly higher prevalence of bronchioloalveolar cell carcinoma and adenocarcinoma has been reported in some series.549. and 552. Because lung cancer is usually a late complication of systemic sclerosis, 413 surgical treatment is rarely possible.
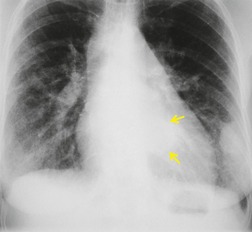
Pneumonia is a recognized complication of systemic sclerosis, and, in some cases, may be an aspiration pneumonia related to esophageal dysfunction. 490 Because of the esophageal dysmotility, there is a possibility that chronic aspiration contributes to lung fibrosis; 553 however, in a study by Troshinsky et al., 554 which evaluated esophageal manometry, acid reflux (by pH probe), and pulmonary function in 39 patients with scleroderma, no relationship was found between the degree of reflux or motility impairment and lung fibrosis. An association between esophageal dysmotility and reduced lung volumes in patients with systemic sclerosis has been reported, but this observation probably reflects simultaneous involvement of the lungs and esophagus, rather than lung damage due to aspiration. 555 Furthermore, clinically obvious aspiration is relatively rare in these patients. Most authors consider that aspiration does not play a significant part in the pathogenesis of basal fibrotic changes. 519
Because of the high prevalence of asymptomatic abnormalities on CT scans in patients with scleroderma, it is important to determine which types of abnormality are likely to progress. Remy-Jardin et al. 532 used CT to follow 17 patients with scleroderma. Five patients with initially normal CT scans, but with abnormal BAL, remained normal on follow-up CT. Six of seven patients whose initial CT scans showed some honeycombing progressed on follow-up evaluation, with new honeycombing in areas previously occupied by ground-glass attenuation. Five patients had abnormal initial CT scans without honeycombing: three of these were unchanged on follow-up, while two patients had resolution of some of their changes. However, a more recent study suggested that both honeycombing and ground-glass abnormality tended to progress. 556 In our experience, patients with moderate or extensive parenchymal abnormality, or those with honeycombing, tend to progress over time, while those with relatively mild ground-glass or reticular abnormality show only minor fluctuation. A very few patients show a sudden catastrophic progression of their lung disease with widespread DAD, 557 presumably similar to that seen in accelerated UIP.
The lung fibrosis associated with scleroderma is associated with a much better prognosis than that found in IPF.35.557. and 558. This is most likely due in part to the predominant NSIP histology. There may also be a component of lead time bias, since early lung fibrosis is often detected in patients with scleroderma because of increased surveillance. Morphologically, patients with lung fibrosis related to scleroderma differ from those with IPF because of a greater degree of upper lobe involvement, and by a coarser reticular pattern, again probably due to the predominance of NSIP. 527
CREST syndrome (limited cutaneous systemic sclerosis)
The CREST syndrome is a clinical subtype of scleroderma in which (C)alcinosis, (R)aynaud’s phenomenon, (E)sophageal dysmotility, (S)clerodactyly, and (T)elangiectasia are prominent features.559. and 560. It is commonly associated with the anticentromere antibody (ACA). 498 Although patients with this syndrome often have less severe skin and visceral involvement than in scleroderma, about 60% of patients with CREST syndrome develop significant pulmonary hypertension. 561 Selection bias in various series makes prevalence difficult to assess, but it is probably similar to that of classical systemic sclerosis. 514 The majority of patients are older women presenting with a long history of Raynaud phenomenon and swollen fingers, but less extensive skin involvement than systemic sclerosis. 514 Between 50% and 80% of patients have ACA – an unusual finding in systemic sclerosis.493. and 514. It also differs from systemic sclerosis in that life expectancy is greater, and the disease is quite often mild and slowly progressive. Systemic involvement such as of the kidney is less. 562 However, the CREST syndrome is by no means benign, and there are several reports of severe pulmonary hypertension and deaths from cor pulmonale.562. and 563. Although earlier reports suggested that the prevalence of pulmonary hypertension in CREST syndrome is about 10%,563. and 564. more recent studies found echocardiographic evidence of pulmonary hypertension in 30–60% of cases.542. and 561. The reported prevalence of lung fibrosis in CREST syndrome has ranged from 0% to 72%, depending at least in part on the criteria used for evaluation518.562.565. and 566. In studies using HRCT, the prevalence of parenchymal abnormality is 25–30%,498. and 533. but the extent of abnormality is less than that seen in diffuse scleroderma. 533
Overlap – systemic sclerosis syndrome
This is the other major clinical variant of systemic sclerosis, constituting 10–27% of all cases of systemic sclerosis.495. and 518. Overlap occurs with one or more connective tissue disorders: SLE, rheumatoid disease, and dermatomyositis. Pleuropulmonary involvement is more common than in classical systemic sclerosis, with radiographic fibrosis in 25–65%495. and 518. and pleural effusions in 15–36%.518.558. and 567.
Polymyositis/dermatomyositis
Polymyositis and dermatomyositis are diffuse inflammatory myopathies of striated muscle. Additionally, in DM there are characteristic skin changes. Several subgroups are identified and these may be classified as in Box 10.27. 568
Box 10.27
• Primary idiopathic polymyositis
• Primary idiopathic dermatomyositis
• Amyopathic dermatomyositis
• Polymyositis or dermatomyositis in association with:
– Neoplasia
– Other collagen vascular disease (overlap syndromes)
• Polymyositis or dermatomyositis of childhood
• Antisynthetase syndrome569
*Adapted from Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med 1975;292:344–347.
Female patients outnumber males about 2 : 1, 331 and most present at between 40 and 60 years of age, with a smaller peak between the ages of 5 and 15. 568 Clinical presentation may be with an acute, subacute, or chronic illness characterized by a progressive, symmetric weakness of the girdle and neck muscles. In the acute disease, muscle pain and tenderness are common, and there may also be pharyngeal and respiratory symptoms. 570 In dermatomyositis, additional and characteristic skin changes are present: a heliotrope periorbital rash, and a violaceous/red papular rash over bony prominences. Associated findings, particularly in the subacute form, include systemic symptoms, arthropathy, dysphagia, pulmonary disease, and cardiac disease (Figs 10.50 and 10.51).570. and 571. Anti-Jo-1 antibodies are commonly present, and may precede the onset of clinical myopathy. Although myopathy is the basis of the diagnosis, 568 there is increasing recognition of amyopathic myositis, where muscle weakness is absent in patients who have otherwise typical manifestations of dermatomyositis/polymyositis, including lung involvement.572. and 573. The antisynthetase syndrome569 refers to a constellation of myositis, inflammatory arthritis, infiltrative lung disease, keratoconjunctivitis sicca, Raynaud phenomenon, mechanic’s hands (lateral erythema and hyperkeratosis), and autoantibodies to tRNA synthetases. 574 While Jo-1 is the most common of these antisynthetase antibodies, several others exist, 575 which seem to preferentially identify individuals with amyopathic lung disease.576. and 577. Infiltrative lung disease is particularly common in the antisynthetase syndrome, occurring in up to 80% of cases, 569 often with a characteristic appearance as described below.
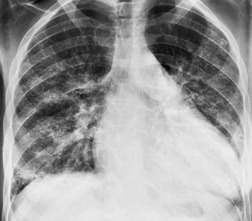
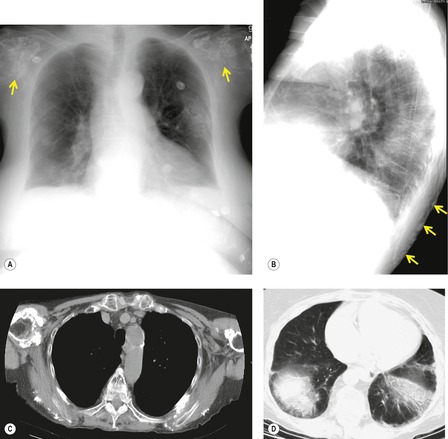
Pulmonary involvement is quite common in polymyositis/dermatomyositis and may occur in up to 50% of patients. It is an important determinant of the clinical course and contributes directly to death in some 10% of patients. 578 The chest manifestations of PM and DM are listed in Box 10.28.
Box 10.28
Primary
• Infiltrative lung disease
– Organizing pneumonia
– NSIP
– UIP
– DAD
• Vascular
– Vasculitis
– Capillaritis/hemorrhage
– Pulmonary hypertension
Secondary
• Muscle weakness
– Respiratory failure
– Pneumonia/aspiration
– Atelectasis
• Drug related disorders
• Heart failure
• Lung cancer
Interstitial lung disease
Interstitial fibrosis was first described in dermatomyositis in the 1950s. 578 It is now a well-recognized association579 that occurs in up to 40% of patients. 580 It is slightly more common with polymyositis than with dermatomyositis. It may also occur in amyopathic dermatomyositis. The presence of ILD in polymyositis/dermatomyositis correlates strongly with the presence of anti-Jo-1.470. and 581. About 50–70% of patients who are anti-Jo-1 positive have ILD571 whereas the frequency of ILD falls to about 10% if antibodies are absent. ILD may antedate myositis in patients with anti-Jo-1 antibodies. 582
ILD tends to be accompanied by joint involvement583 and, as with ILD in other collagen vascular diseases, pulmonary changes on the chest radiograph can be the first clinical manifestation, preceding the skin rash or myositis,583.584. and 585. although the usual pattern is for the ILD to follow soon after the onset of muscle weakness. The clinical manifestations vary greatly. At one extreme is an acute, rapidly fatal illness resistant to therapy571.586.587. and 588. in which the muscle disease can be masked by respiratory involvement. At the other is a benign, indolent, and asymptomatic form.586. and 588. It is said that the ILD of polymyositis/dermatomyositis is more steroid responsive than that of systemic sclerosis, 331 perhaps due to a higher prevalence of organizing pneumonia. About half the patients with lung involvement respond with lessening of dyspnea, clearing of the chest radiograph, and improvement in lung function tests.578. and 587. The variation in the clinical course of ILD in polymyositis/dermatomyositis and its generally favorable response to steroid therapy reflects the variety of underlying pathologic changes and their relative frequency. The most common pathologic findings are NSIP202.573.589. and 590. and organizing pneumonia,591.592. and 593. often occurring in combination. A rapidly progressive form of lung disease is associated with DAD.594.595. and 596.
Characteristic imaging changes commonly consist of symmetric, basally predominant reticulonodular opacity, often with superimposed airspace opacity (Fig. 10.50).586. and 587. Lung volumes are often markedly reduced, particularly in the lower lungs. Although pleuritis, fibrosis, and small effusions are common pathologically, 587 they are uncommon on imaging. The chest radiograph may show muscle calcifications (Fig. 10.51).
Lung disease associated with polymyositis/dermatomyositis or with the antisynthetase syndrome is often associated with a characteristic CT appearance, characterized by confluent ground-glass opacity and consolidation in the lower lobes, superimposed on a background of reticular abnormality with traction bronchiectasis (Fig. 10.52).591.597. and 598. This pattern reflects the characteristic histologic combination of organizing pneumonia and fibrotic NSIP.591. and 598. Parenchymal band opacities, typically basal and peripheral, and irregular thickening of bronchovascular interfaces may be seen. Honeycombing is relatively uncommon, with recorded prevalence ranging from 0% to 26%, and reflects underlying UIP.591.592. and 598. In patients with acute rapidly progressive lung disease due to DAD, extensive consolidation and ground-glass opacity are seen. 591 On serial evaluation, the changes of consolidation, ground-glass abnormality, reticular abnormality, and traction bronchiectasis may all be partially reversible with treatment.590.591. and 597. Less commonly, consolidation may progress to reticular abnormality.
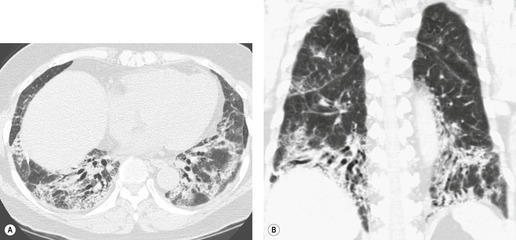
When the diaphragm becomes involved in the myositis599 functional and radiographic changes are produced similar to those seen with the diaphragmatic myopathy of SLE. Characteristically the myopathy produces bilateral hemidiaphragm elevation, reduced lung volume, and discoid basal atelectasis.578. and 599. Pulmonary arterial hypertension571 is relatively uncommon in polymyositis/dermatomyositis, except as a complication of ILD (see Fig. 10.51). 585
Secondary manifestations
Aspiration pneumonia may occur in patients with pharyngeal or esophageal dysfunction due to polymyositis/dermatomyositis, 578 and should be considered in the differential diagnosis of new parenchymal opacities.
Malignancy and polymyositis/dermatomyositis
There is a two- to sevenfold increase in the frequency of malignant disease in polymyositis/dermatomyositis,413.600. and 601. especially in older patients. The average prevalence in various series is 21% for DM and 15% for PM. 602 The carcinomas recorded roughly parallel their frequency in the general population, 602 though ovarian cancer may be more common. 603 Polymyositis/dermatomyositis may precede, accompany, or follow the carcinoma, though it is usually found within 1 year of diagnosis. 604 The course of the myopathy often parallels the course of the malignancy, improving when the malignancy is treated, and worsening with relapse, suggesting that it is a true paraneoplastic syndrome. 605 The prevalence of malignancy in most series is higher in patients with dermatomyositis than in those with polymyositis,604. and 606. and cutaneous necrosis or mucinosis should lead to even higher suspicion for malignancy.607. and 608. Using Callen’s data, 609




Related posts:
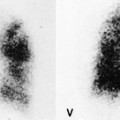
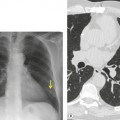
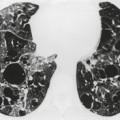
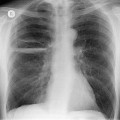
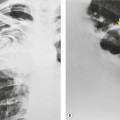
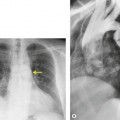
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
