SARCOIDOSIS
Sarcoidosis is a common systemic disease characterized by widespread development of noncaseating epithelioid cell granulomas that eventually either resolve or convert into fibrous tissue. The clinical manifestations of sarcoidosis may be widespread (Box 11.1), or may be restricted to a single organ. However, the lung and its associated lymph nodes are involved in over 90% of cases. Although the cause and pathogenesis of sarcoidosis are obscure, it is thought to be the result of a disordered, immunologically mediated response to one or more unidentified and possibly inhaled agents. Various agents have been suggested, including Mycobacterium species (either alone or with viruses), other infectious organisms, organic antigens (e.g. pollen), and inorganic dusts.1.2. and 3. Given the multiple reported environmental risk factors,4. and 5. it seems most likely that the development of sarcoidosis is probably the end result of immune responses to various ubiquitous environmental triggers. 6 The inciting antigen or antigens trigger a T lymphocyte response that is characterized by chronic inflammation, monocyte recruitment, and granuloma formation. The human leukocyte antigen (HLA) allele DRB1*1101 appears to be a risk factor for sarcoidosis, but the mechanism for this increase in risk is not known. 7 About 1–3% of patients with sarcoidosis have a family history of the disease. 8 The reported occurrence rate for sarcoidosis in first-degree relatives of affected individuals is about 4.6 times that of controls. 9
Box 11.1
Eye
• Iritis
• Conjunctivitis
• Lacrimal gland enlargement
Heart
• Arrhythmia
• Sudden death
• Left ventricular failure
Liver
• Hepatosplenomegaly
• Hepatitis
Nervous system
• Cranial nerve abnormalities
• Focal brain or meningeal lesions
• Pituitary abnormality
Bone
• Polyarthritis
• Lytic bone lesions
Skin
• Erythema nodosum
• Lupus pernio
Other
• Parotid enlargement
• Hypercalciuria, hypercalcemia
Pathology and clinical features
The granuloma is the earliest lesion of sarcoidosis. 10 Early loose granulomas evolve into mature ones that consist of a tightly packed central collection of epithelioid histiocytes and occasional multinucleate giant cells surrounded by lymphocytes (activated T cells), monocytes, and fibroblasts. 1 Necrosis is rare and when it does occur is minimal and confined to the center of granulomas. 11 Granulomas are predominantly interstitial and are distributed along lymphatic pathways, being characteristically found along bronchovascular bundles, interlobular septa, and pleura, 12 with or without interstitial pneumonitis in the adjacent lung. Granulomas, once formed, may remain stable for months or years. They may resolve spontaneously or in response to treatment. In a minority of cases (perhaps 20%) the granulomas become obliterated by centripetal fibrosis that may develop into extensive interstitial fibrosis, with destruction and distortion of lung architecture and the eventual production of an end-stage lung.
A substantial range of disorders can cause granulomatous lung disease (Box 11.2). 13 Distinguishing features of granulomas due to sarcoid or beryllium include their tendency to conglomerate along lymphatic pathways, and the presence of surrounding dense eosinophilic lamellar collagen, which often contains granulomas. 14 They differ from infective granulomas because of the absence of caseous necrosis. The granulomas of hypersensitivity pneumonitis tend to be small, poorly defined, and bronchiolocentric. Intravenous talcosis is characterized by perivascular clusters of giant cells containing birefringent particles. The tendency of sarcoid granulomas to involve other organs is an important feature that distinguishes sarcoidosis from various lung-limited granulomatous disorders. 15 For example, chronic beryllium disease is generally confined to the lungs.
Box 11.2
• Infection
– Mycobacteria
– Fungal
– Metazoa
– Spirochetes
– Other (cat-scratch disease, Whipple disease)
• Chemical/foreign body
– Beryllium
– Silica
– Talc
– Chronic aspiration
• Angiitis and granulomatosis
– Wegener granulomatosis
– Churg–Strauss syndrome
– Necrotizing sarcoidal angiitis
– Bronchocentric granulomatosis
• Pulmonary Langerhans cell histiocytosis
• Hypersensitivity pneumonitis
• Other
– Chronic granulomatous disease
– Lymphomatoid granulomatosis
– Granulomatous response to adjacent cancer or lymphoma
– Immune reconstitution syndrome
– Interferon-α treatment
Incidence rates for sarcoidosis vary greatly from country to country and depend, among other factors, on race, the sophistication of medical care, and use of screening programs. Quoted figures for incidence of clinical disease are of the order of 1–10 cases per 100 000 population per year, but this is almost certainly an underestimate because many cases remain subclinical. 16 Studies performed using mass radiographic screening for tuberculosis have yielded a prevalence of 10–30 cases per 100 000; many of these cases were asymptomatic, and might never have been detected clinically.17. and 18. The highest incidence of this condition is found in northern European countries (5–25 cases per 100 000).17. and 19. In the USA, the annual incidence is about 3.5 times greater in African Americans than in white people. 20 Sarcoidosis begins later in African Americans, and is more likely to be chronic and fatal. 20 In individual cases, the ethnic origin of the patient is relatively unhelpful in determining the likelihood of sarcoidosis.
A number of conditions have been reported to show an association with sarcoidosis; the relationship is best established for tuberculosis. 21 In a series of 425 patients with sarcoidosis, tuberculosis immediately preceded sarcoidosis in 1.6% and sarcoidosis developed into overt tuberculosis in 1.9%. 22 More recently, a sarcoidlike syndrome has been shown to occur as part of an immune reconstitution syndrome in patients treated with highly active retroviral treatment for acquired immune deficiency syndrome (AIDS).23.24. and 25. Sarcoidosis in these individuals may present up to 3 years after the commencement of treatment, and the clinicopathologic characteristics of sarcoidosis in human immunodeficiency virus (HIV)-infected patients are similar to those of sarcoidosis in HIV-uninfected patients. Sarcoidosis may be also related to treatment with interferon-α for chronic hepatitis C26 or melanoma. 27
Sarcoidlike reactions may occur in lung or other tissues adjacent to lymphoma, and lung cancer. 28 Lymphoma (Hodgkin or non-Hodgkin) appears to be the most common associated neoplasm, but lung cancer may also be associated with sarcoidlike reactions. Other associated tumors may include testicular cancer, 29 head and neck cancer, 30 breast cancer, 31 and renal cancer. 32 The relative risk of malignancy in the decade following diagnosis of sarcoidosis is at least twice the expected value, and this is probably due at least in part to initial misdiagnosis of sarcoidosis. 28 The radiologist should be alert for imaging features suggesting neoplasm in individuals with a diagnosis of sarcoidosis.
Sarcoidosis most commonly presents between 20 and 40 years of age,20.33. and 34. but the range is wide, ranging from the first year of life35 to the eighth decade. There is a slight female predominance. 6 The mode of presentation varies greatly among series, depending particularly on racial mix and the use of screening radiography. In studies with predominantly Caucasian populations, presentation as an incidental radiographic finding is common and may occur in 30–50% of cases. 36 Low-grade fever, musculoskeletal symptoms, weight loss, and fatigue are also common. 36 Respiratory illness (21%), erythema nodosum (16%), ocular symptoms (7%), and other skin lesions (4%) 37 represent the other common presentations. In those of black racial origin, respiratory and systemic symptoms such as fatigue, malaise, weakness, weight loss, and fever are most common. 38 A striking difference between black individuals and Caucasians at presentation is in the frequency of erythema nodosum. This finding is uncommon in African Americans but very common in Caucasians, particularly in the UK, where 30–40% of patients present with the condition37.39. and 40. often as part of Löfgren syndrome (erythema nodosum, fever, and hilar adenopathy). 41 In general, sarcoidosis occurring in African Americans is later in onset, is more likely to involve peripheral nodes, skin, and eyes, and to become chronic and disseminated. Because of the lower incidence of the benign Löfgren syndrome and the higher incidence of chronic, disseminated disease, the prognosis of sarcoidosis is worse in African Americans than in white people. 6
Findings on investigation
Hematologic evaluation may show anemia, neutropenia, or lymphopenia. A significant blood eosinophilia has been reported in up to one-quarter of patients.15.42. and 43. Sarcoidosis should therefore be considered in the differential diagnosis of pulmonary eosinophilia, though patients with sarcoidosis do not usually have eosinophilia of the lung parenchyma.
Disordered calcium homeostasis is caused by synthesis of 1,25-dihydroxyvitamin D3 (calcitriol) by activated sarcoid macrophages44 and is rapidly responsive to steroids. 45 Hypercalciuria and hypercalcemia may become prominent features. 1 Hypercalcemia can cause metastatic calcification and renal failure. 33 Hypercalciuria is two to three times more common than hypercalcemia, 46 occurring in about 11% of patients, 37 and may lead to renal calculi.
Serum levels of angiotensin-converting enzyme (ACE), produced by activated macrophages, are raised in about 50–60% of patients.47.48. and 49. Serum ACE levels correlate with the total body granuloma burden. 50 However, measurement of these levels is not diagnostically helpful, as raised levels of serum ACE can also be present in other granulomatous conditions, and in Hodgkin lymphoma.40. and 51. Other biochemical changes may include abnormal liver function (particularly alkaline phosphatase). 50
Various immunologic derangements are seen with sarcoidosis. Cutaneous anergy is well recognized. About two-thirds of patients have a negative tuberculin test, and in many of the remainder it is only weakly positive.37. and 38. Cutaneous anergy is probably related to the relative abundance of suppressor T cells (CD4 cells) in the peripheral blood following sequestration of helper T cells (CD8 cells) in active sarcoid lesions. Activated helper T cells stimulate nonspecific antibody production by B cells, 1 producing a whole range of antibodies and resulting in hypergammaglobulinemia1 in about 50% of patients, particularly in individuals of African races. 37
In active sarcoidosis, bronchoalveolar lavage (BAL) shows T cell lymphocytosis with an increase in activated helper cells, manifest as an increase in the CD4/CD8 ratio38. and 52. and to a lesser extent activated alveolar macrophages. Such lymphocytosis can even be seen in sarcoidosis without evidence of lung disease. 52 This ‘sarcoidosis’ pattern of BAL may also be seen with other conditions such as chronic beryllium disease and hypersensitivity pneumonitis. 52 Although a high CD4/CD8 ratio in BAL is associated with other measures of disease ‘activity’, its usefulness in predicting outcome and monitoring treatment is debatable1 and most clinicians believe it has no role in routine management of the patient with sarcoidosis. 53
Typical physiologic findings in patients with sarcoidosis are restrictive, with a decrease in total lung capacity, a reduced diffusing capacity, and reduced compliance.54. and 55. However, airway obstruction may also be present, either as a manifestation of early disease with small airway involvement56.57. and 58. or in later disease when there is reticular abnormality and bronchovascular distortion. 59 A loose relationship exists between respiratory function tests and chest radiographic changes.55. and 60. The mean values for respiratory function tests correlate with the radiographic stage, becoming worse from stage I to stage III. However, individual exceptions to this correlation are common. For example, several studies have shown that about 50% of patients in stage I have a diffusion defect.55. and 61. Also, respiratory function tests can give results in the normal range in stage II and stage III disease. Discrepancies between radiographs and function are most likely to arise when patients are treated with steroids, which commonly improve function in the face of an unchanging chest radiograph. 55
Diagnosis and prognosis
The diagnosis of sarcoidosis requires clinical and imaging features compatible with the diagnosis, and the identification of noncaseating granulomas in at least one organ. 62 In clinical practice the organs most commonly sampled are lymph nodes, liver, and lung. Transbronchial biopsy is usually the investigation of first choice, with a diagnostic yield of about 90%, provided that enough tissue samples are obtained.63.64.65.66. and 67. Even in patients with hilar adenopathy who have normal lung parenchyma on chest radiographs, the yield from biopsy is over 80%. 68 The high yield of transbronchial biopsy in this condition is due to the predominantly peribronchovascular distribution of the granulomas.
A large number of conditions can produce granulomatous lesions (Box 11.2), and if the diagnosis of sarcoidosis rests on histologic appearances, these other conditions must be excluded as far as possible by history, special histologic stains, and microbiologic culture. Establishing the widespread nature of the granulomatous process is also important, since local sarcoidlike reactions can be produced by a number of primary processes, including lymphoma and carcinoma.69. and 70. In clinical practice, the diagnosis of sarcoidosis is sometimes accepted without biopsy, provided that clinical, laboratory, and imaging features are typical. In particular, biopsy is usually unnecessary when the patient presents with Löfgren syndrome (erythema nodosum, fever, and bilateral hilar adenopathy), and the resolution is rapid and spontaneous. 62 When the clinical diagnosis is less firmly based, histologic proof is essential. The Kveim test, in which skin biopsy is performed 4–6 weeks after intracutaneous injection of a saline suspension of human sarcoid tissue, 71 is now rarely used, largely because the injected material is poorly standardized and is not widely available. 62
The term ‘activity’ is frequently used in relation to sarcoidosis but it is not exactly defined and its clinical relevance is unproven. Current understanding of the pathogenesis of sarcoidosis suggests that an initiating event (antigen exposure) sets in train a three-phase response: (1) T-lymphocyte/macrophage inflammation; (2) granuloma formation; (3) active fibrosis – any or all of which can be active, i.e. changing or evolving. 72 Different markers of activity reflect different elements of this process. A great variety of markers have been proposed, using clinical parameters, imaging tests (chest radiography, computed tomography [CT], gallium-67 scanning), serum levels (e.g. calcium, ACE), and BAL markers. Despite this great variety the consensus view is that the clinical activity of sarcoidosis is best assessed on the basis of the onset, worsening, or persistence of symptoms related to sarcoidosis. 62 In general, tests to stage activity can be limited to clinical investigation, chest radiography, and lung function testing. 72 The basic problem with all markers of disease ‘activity’ is that they do not necessarily predict outcome or the necessity to start corticosteroid therapy. 72
Indications for treating chest disease are not firmly established but most patients with symptomatic or progressive stage II or stage III disease are treated. The clinician will usually base treatment decisions on the patient’s symptoms, physiologic impairment, or evidence of extrathoracic involvement rather than solely on the chest radiographic appearance. If treatment of sarcoidosis is indicated, then corticosteroids are the agents of first choice.10.53. and 73. There is no doubting the short-term efficacy of steroids but their long-term value is less clear and a number of controlled studies have shown no long-term difference between treated and untreated patients74 and symptoms will commonly recur when treatment is reduced or stopped. 62 Other agents have been tried in steroid-resistant sarcoidosis including immunosuppressive, cytotoxic, and antimalarial drugs. 53
Lung transplantation has been successfully employed in end-stage sarcoidosis with survival rates at 1 year that are slightly less than in other forms of diffuse lung disease. 75 Recurrence of sarcoidosis has been described in the transplanted lung76. and 77. but is not usually associated with a significant clinical problem and can be controlled with augmented immunosuppression. In a multi-institutional study, sarcoidosis was found to have recurred in nine (35%) of 26 patients transplanted for this disease, and was the most common disease to recur in transplanted lungs. 78 Recurrent sarcoidosis was identifiable on CT in only 3/9 cases identified histologically. In those with abnormal CT, recurrence was manifested by either miliary nodules (n = 2) or a solitary pulmonary nodule (n = 1). 78
The majority of deaths from sarcoidosis are related to pulmonary, cardiac, or neurologic disease. In three series of 113 sarcoid-related deaths, 69 (61%) resulted from pulmonary involvement – overwhelming granulomatous disease, pulmonary fibrosis, cor pulmonale, hemorrhage, mycetoma formation, and respiratory failure.79.80. and 81. Symptomatic cardiac involvement appears to have a particularly poor prognosis, with mortality up to 20% for symptomatic patients. 82 In several large series, overall mortality ranged between 2.2% and 7.6%.37.62. and 83.
Staging
Sarcoidosis is commonly staged according to its appearance on the chest radiograph. A widely used classification is the following:34. and 84.
• Stage 0 – normal chest radiograph
• Stage I – nodal enlargement only
• Stage II – nodal enlargement and parenchymal opacity
• Stage III – parenchymal opacity without adenopathy or evidence of fibrosis
• Stage IV – lung fibrosis (parenchymal distortion, lobar volume loss, bullae).
Other classifications85 use a three-stage system, with fibrotic and nonfibrotic opacities grouped together as stage III. The use of the term ‘stage’ should not be taken to mean that sarcoidosis always begins as stage 0, with orderly progression through the higher stages. Nevertheless, patients commonly pass sequentially from stage I to stage III. At the time of presentation the percentages of patients at each stage are:
• Stage 0: 5–15%
• Stage I: 45–65%
• Stage II: 30–40%
• Stage III: 10–15%
• Stage IV: 5%.
The stage at presentation is generally considered to correlate with prognosis. Untreated patients with stage I disease experience resolution of symptoms and radiographic abnormalities in 50–90% of cases, compared with 30–70% for stage II, 10–20% with stage III, and 0% with stage IV. 53
Imaging patterns
Normal chest radiograph (radiographic stage 0)
A normal chest radiograph, without adenopathy or parenchymal abnormality, is seen in about 10% of patients with sarcoidosis, depending on the population being studied. 60 Patients with this presentation of sarcoidosis usually have significant extrathoracic disease such as uveitis or skin abnormalities. However, 20–30% of those with normal chest radiographs will have decreased vital capacity and/or decreased lung diffusing capacity. 60 CT may be helpful in clarifying the presence or absence of lung disease in these patients. Of 69 patients with sarcoidosis described in three papers,86.87. and 88. 14 had normal chest radiographs at the time of CT. Of these, seven patients had pulmonary parenchymal disease demonstrable on CT scanning, while the remainder had normal parenchyma on CT. As with other interstitial diseases, a normal chest radiograph or chest CT does not exclude pulmonary sarcoidosis.
Mediastinal and hilar lymphadenopathy (radiographic stage I)
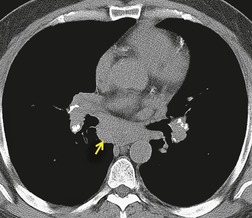
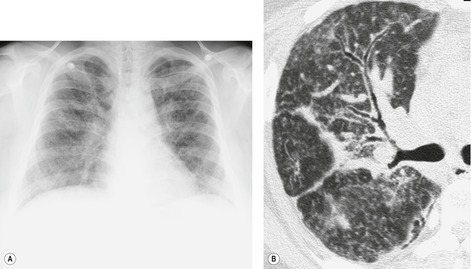
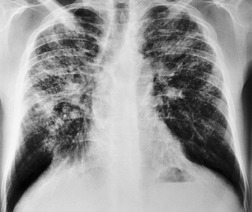
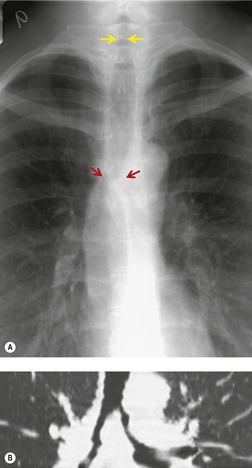
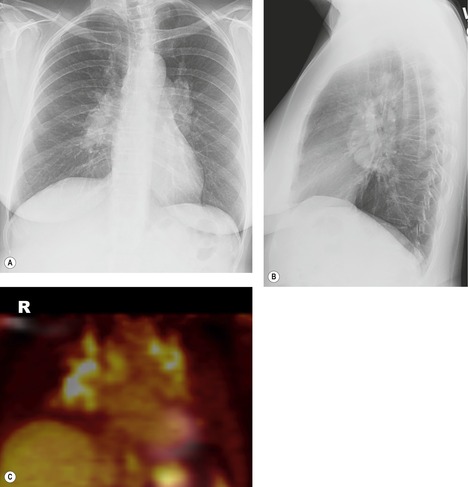
Lymphadenopathy is the most common intrathoracic manifestation of sarcoidosis, occurring in 75–80% of patients at some point in their illness.33.37. and 83. Lymph node enlargement (Box 11.3) is usually seen in the right paratracheal, aortopulmonary window, hilar, and tracheobronchial regions (Fig. 11.1). In one series of 150 patients with sarcoidosis and an abnormal chest radiograph, about 30% had bilateral hilar lymphadenopathy (BHL) alone, 30% had BHL with right paratracheal adenopathy, and 30% had BHL with bilateral paratracheal adenopathy. 33 The aortopulmonary nodes are enlarged in most patients, producing a characteristic local convexity in the aortopulmonary window (Fig. 11.1). 89
Box 11.3
• Hila
• Right paratracheal
• Aortopulmonary
• Subcarinal
• Less common sites of adenopathy
– Anterior mediastinal
– Posterior mediastinal
– Paracardiac
– Retrocrural
– Axillary
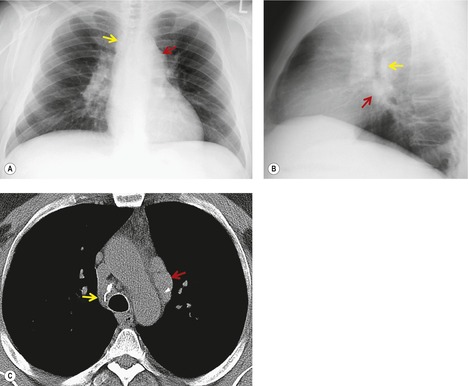
The degree of hilar node enlargement ranges from barely detectable to massive, in which case nodes may reach halfway to the chest wall. The outer margins of the hila are usually lobulated and well demarcated except when there is adjacent parenchymal opacity. 90 Both the more proximal tracheobronchial and the more distal bronchopulmonary nodes are involved in sarcoidosis, and enlargement of the latter is characteristic. 91 These more peripheral nodes may have lung or lower lobe bronchus on their medial aspect, and thus when enlarged they typically have a clearly demarcated inner border (Fig. 11.1). 91 Symmetry is an important diagnostic feature of the hilar adenopathy associated with sarcoidosis because symmetric adenopathy is unusual in the major diagnostic alternatives (Box 11.4) such as lymphoma, tuberculosis, and metastatic disease (Fig. 11.2). Sometimes the adenopathy appears more marked on the right on a frontal radiograph simply because the right hilum is more clearly separated from the mediastinum than the left.
Box 11.4
• Sarcoidosis
• Infection
– Tuberculosis (especially HIV-positive)
– Fungal infection
– Brucellosis
– Tularemia
– Plague
– Anthrax
– Infectious mononucleosis
– Cat-scratch disease
– Viral or mycoplasma pneumonia (mainly in children)
• Neoplasm
– Lymphoma
– Metastases
– Leukemia
• Occupational
– Silicosis
– Chronic beryllium disease
• Other
– Amyloidosis
– Drugs (phenytoin)
– Immune reconstitution syndrome
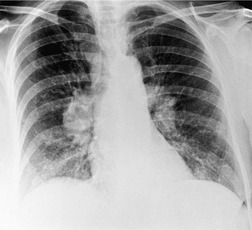
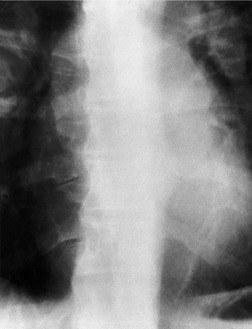
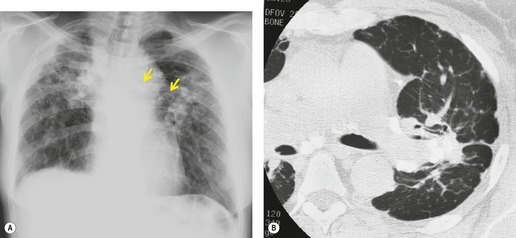
Unilateral hilar adenopathy occurs in 3–5% of cases of sarcoidosis33.90. and 92. (Fig. 11.3). In one series of 135 patients an exceptionally high proportion of patients (9.6%) showing unilateral hilar adenopathy was recorded93 and adenopathy was found to be unilateral in 29% of 21 patients aged 50 years or more. 94 Unilateral hilar adenopathy is about twice as common on the right as on the left, and can occur either alone or with right paratracheal adenopathy.93. and 95. In these patients, more aggressive investigation is required, because the differential diagnosis includes not only malignancy but also primary or AIDS-related tuberculosis or fungal infection.
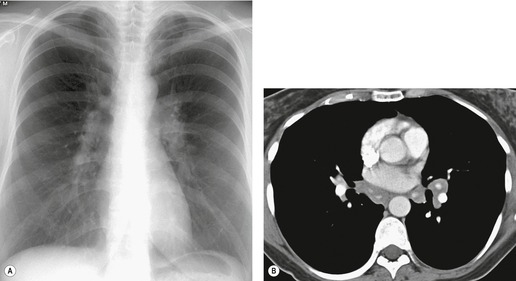
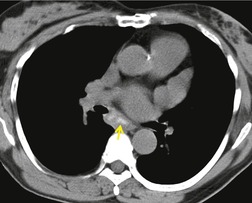
In general, involvement of anterior or posterior mediastinal lymph nodes accompanies the more classic adenopathy, and the degree of enlargement is usually less marked. Although CT scanning commonly demonstrates adenopathy in ‘atypical’ locations such as the anterior or posterior mediastinum, axilla, retroperitoneum, or internal mammary chain, this adenopathy is almost invariably associated with extensive middle mediastinal and hilar adenopathy. 12 Anterior mediastinal nodes are difficult to appreciate on plain radiographs, unless they are calcified (see Fig. 11.7 below), but CT shows involvement in 25–66% of patients (Fig. 11.4).96. and 97. Isolated anterior mediastinal adenopathy, however, is rare90.98.99. and 100. and should strongly suggest a diagnosis other than sarcoidosis, particularly lymphoma.
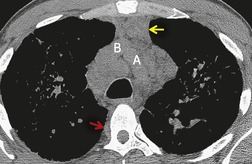
Subcarinal adenopathy occasionally may be marked and can cause airway compression101 or dysphagia. 102 The prevalence of subcarinal adenopathy in a study using chest radiographs was 21%, 89 but with CT (Fig. 11.5) the frequency is in the order of 50%. 96 Involvement of subcarinal nodes in the absence of hilar adenopathy is rare but has been recorded. 103 Posterior mediastinal adenopathy (Fig. 11.6) is one of the least common patterns in sarcoidosis. Only one case report exists of sarcoidosis presenting with isolated posterior mediastinal adenopathy. 104
Natural history of stage I sarcoidosis
The enlarged nodes of sarcoidosis are usually at their maximum size when first seen; they decrease in size over the next 3–6 months so that two-thirds are no longer visible after 1 year; 91 and very few are visible after 2 years. 84 In one review only 6% persisted at 2 years, and most of these were smaller than when first examined. 84 If nodes are still present at 2 years, they commonly remain unchanged for many years91. and 105. and probably indefinitely. Given this fact and the commonness of sarcoidosis, the possibility that mediastinal lymphadenopathy is caused by sarcoidosis must always be considered even in the context of malignant disease.106. and 107.
Of patients with stage I disease at initial examination, about 60% go on to complete resolution,84.91.92. and 93. with resolution being more common in those with erythema nodosum and arthropathy. 84 In the remaining 30–40% of patients presenting with adenopathy, parenchymal opacity develops,84. and 91. usually within the first year, although longer intervals are not uncommon91 and gaps of 10 years or more have been recorded. 105 The adenopathy commonly begins to shrink at the time that parenchymal abnormality appears,33. and 84. a feature that can be helpful in distinguishing sarcoidosis from lymphoma or carcinoma, in which the nodes would usually enlarge in parallel with progressive pulmonary involvement. 93
Fluctuation of nodal enlargement during intermittent steroid therapy is well recognized, 108 but once the nodes have completely resolved on imaging they probably will not enlarge again spontaneously. Such a train of events should raise the possibility of malignant adenopathy. 109 Only about 10 cases of nodal recurrence have been reported, and these spanned periods of up to 20 years. 110 Nodal recurrence developed either alone41. and 108. or with other features of sarcoidosis. 111 Recurrences may be multiple, and as many as three or four episodes have been recorded. 84 Different nodal groups may be involved in separate episodes, as in one patient in whom first one hilum and then the other was involved. 112
Nodal calcification
The frequency of detection of nodal calcification in sarcoidosis (Fig. 11.7) is related to the mode of detection and the duration of disease. 113 In short-term studies using chest radiography, nodal calcification is seen in about 1–3% of cases,33. and 84. but in a longer term study of 111 patients observed for 10 years or more the prevalence was 20%, 114 leading to the suggestion that sarcoid is probably the most common cause of mediastinal nodal calcification in regions where the prevalence of tuberculosis or fungal infection is low.
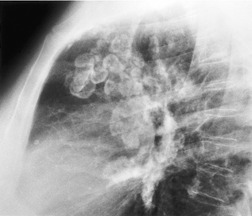
The occurrence of calcification is unrelated to either hypercalcemia or coincident tuberculosis. Most descriptions are of paratracheal and hilar involvement, but any nodal group may be involved. 115 The morphology of the calcification on chest radiographs is variable and nonspecific. However, in a number of patients, peripheral, eggshell calcification develops (see Fig. 11.7).90.99.115.116. and 117. This is of diagnostic value because its occurrence is largely limited to sarcoidosis and silicosis. 117 Spontaneous resolution of eggshell calcification has been reported. 22
CT allows a more detailed analysis of the frequency and morphology of nodal calcification than the chest radiograph. The prevalence of nodal calcification detected by CT in patients with sarcoidosis is from 45%118 to 55%. 116 Gawne-Cain and Hansell116 compared findings in 28 patients with tuberculosis and those in 49 with sarcoidosis, selected on the basis that they had had mediastinal CT. In sarcoidosis, the number of calcified nodes is greater, the calcified nodes are larger, and hilar calcification when present is more commonly bilateral. Nine percent of sarcoid nodes showed an eggshell pattern and in 4% this was the predominant pattern. In other patients with sarcoidosis, a central, less dense, cloudlike or icing sugar pattern of nodal calcification is seen (Figs 11.5 and 11.8), differing from the punctate dense calcification typically seen in tuberculosis or fungal infection.
Parenchymal sarcoidosis (stages II–IV)
Parenchymal opacity is seen radiographically at the time of presentation in a little under half of the patients with sarcoidosis.33.37.60.92.93. and 119. In addition, about one-third of patients presenting with stage I disease go on to develop parenchymal opacity, with a frequency that shows a marked variation from series to series, ranging from 10% to 43%.15.84.92.93. and 119. Such parenchymal abnormality frequently develops within the year and is usually accompanied, at least to some degree, by nodal regression. Parenchymal abnormalities on the chest radiograph or chest CT in sarcoidosis may usefully be classified into reversible and irreversible opacity.
Potentially reversible changes
Potentially reversible changes on the chest radiograph in sarcoidosis consist of three patterns: reticulonodular opacities, ill-defined opacities with the characteristics of consolidation (alveolar), and large nodules. These patterns can occur alone or in varying combinations. 33 They may resolve partially or completely, or they may progress to an irreversible, fibrotic pattern. The approximate frequency of the various patterns of parenchymal abnormality is reticulonodular, 75–90%; ‘alveolar’, 10–20%; and large nodular, 2%.
Reticulonodular and nodular opacities
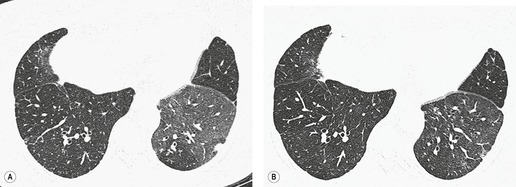
Small, rounded, or irregular opacities constitute by far the most frequent pulmonary pattern and are seen in some 75–90% of patients with parenchymal abnormalities.60. and 120. Irregular or reticulonodular opacities are more common than pure nodules. 60 The nodules range in size from just under 1 mm to over 5 mm, with most 2–4 mm in diameter (Fig. 11.9, Fig. 11.10 and Fig. 11.11).33. and 120. In addition to discrete interstitial abnormalities such as nodules, more diffuse changes occur and are seen on radiographs as bronchial wall thickening, air bronchograms, and subpleural and fissural thickening (Fig. 11.10). 121 Aggregated subpleural granulomas account for the fissural thickening and these can be well appreciated on CT (see Fig. 11.13).
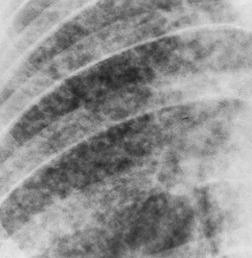
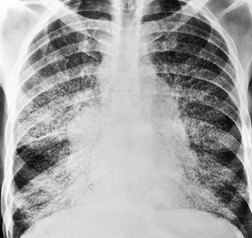
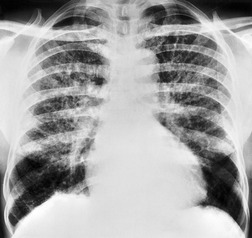
The reticulonodular abnormality is usually bilaterally symmetric (Figs 11.10 and 11.11), with about 15% showing significant asymmetry. 84 In exceptional cases (less than 1% of cases)93. and 122. parenchymal changes are strictly unilateral. Unilateral change, however, was recorded in 8% in one series. 120 Unilateral parenchymal disease is usually, but not exclusively, of the (reticulo) nodular type and may occur with or without adenopathy.113.123.124. and 125.
Opacities tend to occur in all zones, often showing a middle zone or middle and upper predominance (Fig. 11.11).84. and 119. Isolated lower zone involvement is seen in less than 4% of patients, 120 mimicking idiopathic pulmonary fibrosis. Calcification within the lung in parenchymal sarcoidosis is rarely reported. It developed radiographically in five of 136 patients in one series followed for at least 5 years126 and in three of 111 cases followed for 10 or more years. 114 Calcification is more commonly detected on CT.
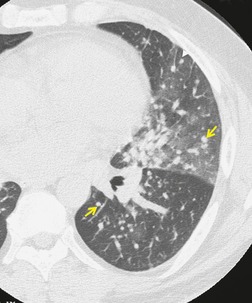
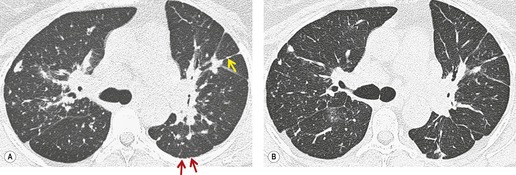
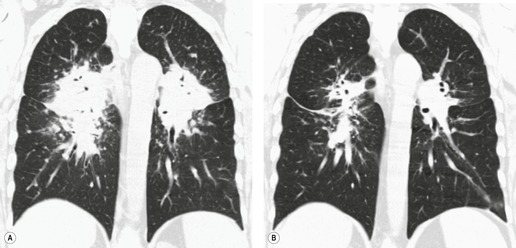
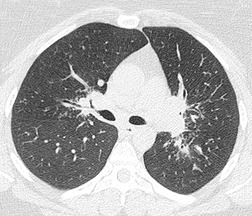
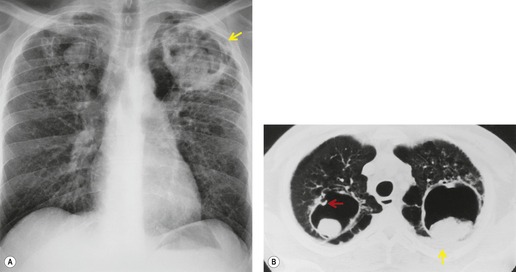
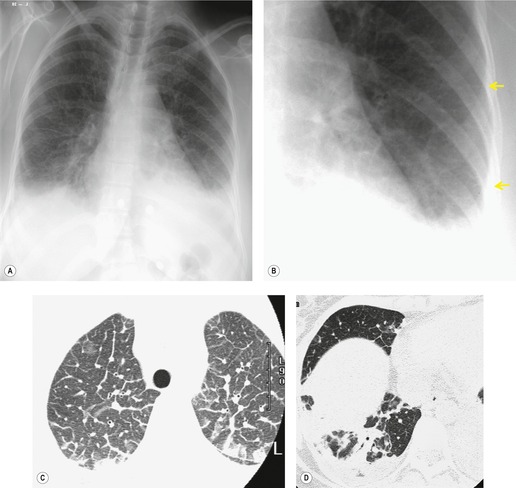
On CT, the nodular or reticulonodular pattern of sarcoidosis is characterized by peribronchovascular thickening and irregularity with small nodules distributed in a perilymphatic fashion (Figs 11.12 and 11.13). 127 The craniocaudal distribution of this abnormality varies, but it often shows a mid- to upper lung predominance. Several studies correlating histology and CT appearances have confirmed the predilection for sarcoid granulomas to lie in or adjacent to lymphatic vessels.12.88. and 128. Because of this characteristic distribution, the nodules are found: along bronchovascular margins (Fig. 11.13, Fig. 11.14 and Fig. 11.15), along interlobular septa (Fig. 11.15), and subpleurally (Fig. 11.15; Box 11.5). 129 The subpleural nodules are often particularly well seen along the fissures (Figs. 11.13 and 11.16). Nodules may also be centrilobular (Figs 11.14 and 11.16). 130 Nodules abutting interfaces make the normally sharp borders of vessels, airways, pleura, and septa irregular and beaded. The overall number of nodules varies greatly, ranging from marked total lung involvement (often upper and middle zone predominant)86. and 131. to a scanty, focal distribution. 88 The nodules typically measure 1–5 mm, but conglomeration into larger irregular nodules or masses is quite common. They are usually sharply defined. A nodule-predominant pattern may resolve completely on CT in about 40% of cases. 132
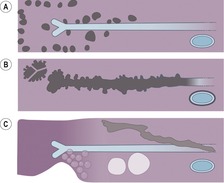
Box 11.5
• Peribronchovascular
• Interlobular septa
• Fissures
• Pleura
• Centrilobular
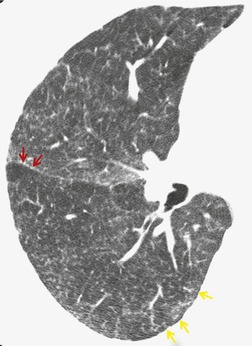
A perilymphatic distribution of nodules may also be seen in lymphangitis carcinomatosa133.134.135. and 136. and lymphoproliferative disease.137. and 138. Lymphangitis carcinomatosa may usually be differentiated from sarcoidosis by the presence of dramatic septal thickening outlining the secondary pulmonary lobule, much more marked than is usually seen in sarcoidosis. 139 Although septal lines are present in about 50% of patients with sarcoidosis, they are not profuse and do not usually form polygonal structures.12.86. and 140. Septal thickening in sarcoidosis may be associated with lobular distortion, a finding not seen in lymphangitis carcinomatosa. Lymphoproliferative disease may also present with predominantly peribronchovascular nodules, but these are usually larger and less profuse than those seen in sarcoidosis. 139
A reticular pattern is an uncommon manifestation of sarcoidosis, but has been described on CT in two patients, who did not have nodules, and had an overall appearance suggestive of idiopathic pulmonary fibrosis141 and quite unlike sarcoidosis. Probably the commonest type of linear opacity in sarcoidosis is thick, nonseptal irregular lines caused by scarring (Fig. 11.15). These are reported in about 40% of patients (range 22–72%).12.118.131. and 142. Occasionally, curvilinear subpleural lines may be identified. 131
Ground-glass opacity
Ground-glass opacity is much more easily seen on CT than on the chest radiograph. This abnormality (Figs 11.14 and 11.17) is seen on CT in about 40% of patients with sarcoidosis (range 16–83%).86.118.131.142. and 143. It is patchy and may have a lobular distribution. In early reports of the CT findings of sarcoidosis the ground-glass opacity was ascribed to alveolitis88 but it is now thought to be due to the presence of innumerable small interstitial granulomas, beyond the resolution of CT.128.144.145. and 146. It is usually, but not always, reversible with steroid treatment,118.144. and 147. with resolution being more likely if it is of short rather than long duration. 142 However, in one long-term follow-up study, an initial ground-glass pattern decreased in extent but evolved into a honeycombing pattern in 4/5 patients. 132
Alveolar and large nodular opacities
In some patients with sarcoidosis, opacities with consolidative (airspace) features develop. These opacities form a spectrum ranging from ill-defined, irregular opacities of nondescript shape (Fig. 11.18) to those that are focal, nodular, and quite well defined. The pathologic basis for the alveolar pattern in sarcoidosis is loss of alveolar air because of compression of the alveoli by coalescent granulomas.90.148. and 149. Histologic study may additionally show alveolar filling with macrophages150 or granulomas.90. and 151. Rarely, patients with bronchial occlusion by endobronchial granulomas may exhibit alveolar opacities due to obstructive pneumonitis. 149
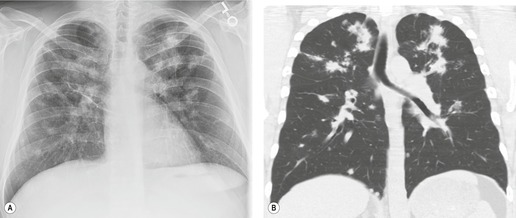
Alveolar sarcoidosis has been reported to have a prevalence rate of 10–20%, but now appears less common.33.90.151.152. and 153. The typical appearance is bilateral, multifocal, poorly defined opacities,33. and 101. ranging in size from about 1 cm to 10 cm. 90 These opacities can occur anywhere, but they show a predilection for the mid-lung zone.90.151. and 152. They may be peribronchovascular or subpleural. An air bronchogram is commonly present.33.101. and 152. At the edge the lesions often break up into a nodular pattern that may be very fine, creating an appearance of acinar rosettes. Reticulonodular opacities are also present in about two-thirds of these patients,33. and 152. and their detection provides a helpful clue when the principal radiographic finding is multifocal consolidation of obscure origin. Another helpful pointer is mediastinal adenopathy, which is an even more common accompaniment, occurring in more than 80% of cases.33. and 152. Kirks and co-workers33. and 154. point out that all of their patients with alveolar sarcoidosis had mediastinal adenopathy or reticulonodular abnormality or both. CT of patients with alveolar sarcoidosis usually shows irregular masslike opacities (Fig. 11.18), with the characteristic peribronchovascular or subpleural distribution. 155 Air bronchograms are often seen, but the irregular well-defined border of the masses serves to distinguish these opacities from true consolidation.86. and 146.
Recognized variants of the alveolar pattern include unilateral distribution, 21 upper zone distribution simulating tuberculosis, 125 or a peripheral pattern that resembles cryptogenic eosinophilic pneumonia or cryptogenic organizing pneumonia. In one series of 64 patients, 9% showed a pronounced peripheral distribution, some with an alveolar and some with a reticulonodular pattern. 156 Sharma153 describes two confusing cases in which the peripheral alveolar pattern was accompanied by blood eosinophilia, yet lung biopsy showed noncaseating granulomas consistent with sarcoidosis. Peripheral consolidation is usually accompanied by nodal enlargement, but exceptions are recorded. 157 Some authors have stressed the good prognosis and tendency for rapid clearing with alveolar sarcoidosis; in one report the opacity cleared in 21 of 22 patients with or without steroids, 152 and in another, clearing occurred in 66% of patients within 9 months. 151 However, in a different study, clearing occurred in only 31%, 33 and in a recent CT-based study, an initial consolidative pattern evolved into a honeycombing or linear pattern in all three of three patients. 132 Relapses may occur. 158 Patients with alveolar sarcoidosis usually have relatively minor physiologic impairment, and have a low prevalence of extrathoracic sarcoidosis. 151
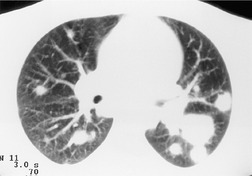
Focal opacities with the imaging features of large nodules (>1 cm diameter) are an uncommon but well-recognized finding in sarcoidosis (Figs 11.19 and 11.20), and when the various series are combined a 2.4% prevalence rate (35 of 1449 cases) is seen.21.33.92.152. and 159. On CT scanning of the large nodular form of sarcoidosis, the nodules may exhibit an irregular outline, with innumerable adjacent small satellite nodules, a finding termed the CT galaxy sign. 160 Patients with the nodular pattern on CT tend to have relatively mild physiologic impairment. 161 The nodules typically resolve completely with treatment. 132
Irreversible fibrotic changes
Sarcoid granulomas may resolve completely, or they may heal by fibrosis (Fig. 11.21). Such fibrosis ranges from being minor and radiographically undetectable119 to marked scarring and distortion (Fig. 11.22). Gross fibrosis is present on the initial chest radiograph in about 5–25% of patients.33. and 148. In addition, gross fibrosis develops in 10–15% of patients who, when first examined, have stage 0 to stage II disease.101. and 119. The fibrosis usually takes years to develop, 101 with a range in one series of 2–14 years. 119
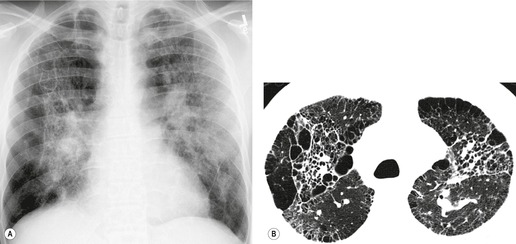
The imaging changes of fibrosis in sarcoidosis are fairly characteristic. 84 The findings classically consist of permanent, coarse, linear opacities radiating laterally from the hilum into the adjacent upper and middle zones, often associated with upward hilar retraction and vascular and fissural distortion (Fig. 11.21).33.90. and 93. The fibrosis is often associated with conglomerate parahilar opacities in the middle and upper zones that resemble those seen in progressive massive fibrosis (Fig. 11.22).84.96. and 119. Coincident with the development of scarring, the extreme upper and lower zones tend to become transradiant (Fig. 11.21). In the upper zones this transradiancy is the result of cyst and bulla formation, whereas in the lower zones it is more commonly the result of compensatory hyperinflation following upper zone volume loss. Cor pulmonale may supervene in the fibrotic stage162 and may be recognizable on imaging. 163
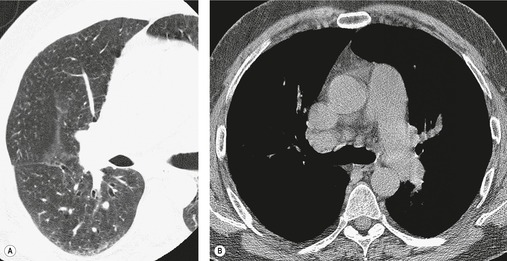
CT has helped to characterize the findings of lung fibrosis in sarcoidosis. Typically one sees areas of conglomerate fibrosis, usually centered on the upper lobe bronchi and vessels, and often associated with marked bronchovascular distortion, characterized by posterior displacement of the main and upper lobar bronchi. 86 These masslike lesions (Figs 11.22 and 11.23) are similar to the progressive massive fibrosis seen in pneumoconiosis, though the masses of sarcoidosis tend to be more perihilar than those of progressive massive fibrosis. The conglomerate masses may decrease in size over time, but bronchial distortion tends to increase (Fig. 11.23). 132 Other signs include lobular distortion, found in about 50% of patients,12.86.118. and 144. long, irregular, linear opacities; traction bronchiectasis and bronchiolectasis; honeycombing and cyst or bulla formation (Figs 11.24 and 11.25).
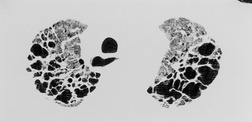
Honeycombing is less common in sarcoidosis than in other forms of end-stage lung disease; the honeycomb cysts tend to be larger than in idiopathic pulmonary fibrosis164 (Fig. 11.26). The honeycombs and cysts of sarcoidosis tend to be distributed subpleurally in the mid- and upper zones of the lungs, sparing the bases. 165 The pathogenesis of honeycombing in sarcoidosis remains unexplained.
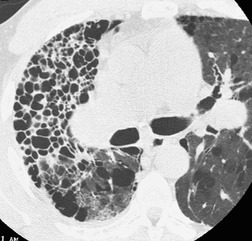
Cavitation and cystic change
The prevalence of cavitation or cystic change in sarcoidosis has been estimated at 2.2%. 166 Cavitation most often occurs in areas of advanced fibrosis, but sometimes in alveolar or large nodular sarcoidosis (Fig. 11.27). Complications such as hemoptysis or aspergilloma occur in about 35% of individuals with cavities. 166 Cavitation is commoner in necrotizing sarcoidal angiitis (see Chapter 10, p 618) and this diagnosis should be considered particularly if the lesions are peripheral and pleurally based.
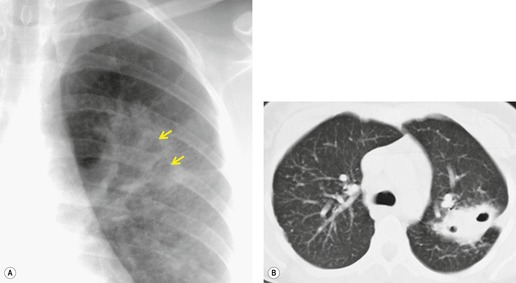
Airway involvement
Sarcoidosis can affect the trachea, bronchi, or bronchioles (Box 11.6). Tracheal involvement is rare167 and in the few reported cases has been associated with laryngeal involvement. Both the proximal and distal trachea may be affected, and the stenosis can be smooth,168. and 169. irregular and nodular (Fig. 11.28), 170 or even masslike. 171
Box 11.6
• Tracheal or bronchial granulomas
• Bronchial stenosis
• Bronchial distortion
• Small airways abnormality with air-trapping
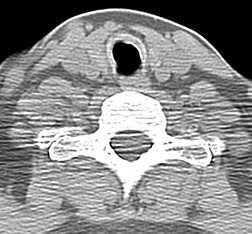
Despite the sometimes massive enlargement of nodes in sarcoidosis, lymphadenopathy alone is a rare cause of symptomatic airway narrowing, 172 perhaps because the nodal capsules remain intact so that scarring and fixation are rare. 113 However, the parahilar conglomerate fibrosis of sarcoidosis commonly causes bronchial distortion and narrowing, and is well shown on CT.
Although endobronchial stenoses (Figs 11.29 and 11.30) occur most commonly in well-established disease, 84 often with pulmonary fibrosis, they have been recorded early in the course of sarcoidosis and with stage 0 radiographic disease.173. and 174. The frequency of large airway narrowing is about 5% (range 2.5–9%).84.119. and 174. Patients present with wheeze, stridor, or airflow limitation173. and 175. or episodes of lobar or segmental collapse and consolidation. Collapse is a well-recognized but uncommon manifestation of sarcoidosis, occurring in about 1% of cases.33.90.92.93. and 148. Any lobe may be affected, most commonly the middle.92. and 113. Collapse of a whole lung has been recorded.
Although the middle lobe is the most common to collapse, stenoses are distributed throughout the lung and show no particular lobar or segmental predilection. The stenoses may be single84. and 174. or multiple,173.174. and 175. and most commonly affect lobar or proximal segmental divisions (Fig. 11.30). At bronchoscopy, the mucosal lesions may be granular, hyperemic, edematous, or masslike.94.176. and 177. With fibrotic lesions, the stenosis may be associated with normal-appearing mucosa. 173 The stenoses may clear spontaneously or with steroid treatment, 178 or may remain unchanged or progress. 174 Mechanical dilatation or stenting is a therapeutic option.179. and 180. When bronchial stenosis is being sought, contiguous thin section acquisition through the central bronchi, with multiplanar reconstructions, is important (Fig. 11.29). 181
The extent of airway involvement by sarcoidosis is underestimated by the chest radiograph. In a CT study of airways in 60 patients with sarcoidosis, 65% had nodular or smooth thickening of the airways from lobar to subsegmental level. 182 Twenty-three percent of patients had luminal narrowing, either smooth or irregular. Most of the patients in this study had stage II disease and the presence of bronchial wall thickening at CT correlated with biopsy-proven bronchial granulomata. Endobronchial sarcoidosis was associated with a high prevalence of airway hyperreactivity in one study, even in the absence of significant airway narrowing. 183
The occurrence of small airways disease in sarcoid patients was recognized by Carrington in 1976. 184 He described the deposition of peribronchial granulomas which result in air-trapping at the level of the secondary pulmonary lobule. This finding was demonstrated by CT 20 years later185 and since confirmed by multiple investigators.59. and 186. Small airway obstruction is seen on expiratory high-resolution CT (HRCT) as air-trapping and patchy areas of low attenuation. 185
Airflow obstruction is common in sarcoidosis.54.187. and 188. Potential etiologies include small airway obstruction, endobronchial narrowing, 189airway hyperreactivity, 183 and bronchial distortion by masslike fibrosis. CT evaluation can be helpful in discriminating among these possibilities. Hansell et al., 59 in a study of 45 patients with airway obstruction in sarcoidosis found that a reticular pattern (presumably associated with bronchial distortion) was the major determinant of airflow limitation, while Davies et al. 58 found that the extent of air-trapping on expiratory CT correlated well with physiologic evidence of airway obstruction. However, a study from Japan suggested that cigarette smoking accounted for most of the expiratory airflow obstruction identified in sarcoid. 190 Airflow obstruction in sarcoidosis has also been shown to be associated with thickening of bronchovascular bundles on CT. 191 These studies confirm that physiologic evidence of airway obstruction is to be expected both in patients with advanced fibrotic sarcoid and in patients with earlier disease who have small airway obstruction.
Pulmonary vascular involvement
Large vessel involvement is rare in sarcoidosis192 and is usually the result of compression by large nodes, or bronchovascular distortion by conglomerate masses.193.194.195.196. and 197. Typically the lobar divisions, particularly the upper lobe vessels, are involved, and pulmonary arterial hypertension is usually absent. Small vessel involvement by granulomas is common and is usually accompanied by widespread parenchymal disease.68. and 184.
Pulmonary artery hypertension is found in 6–23% of patients with sarcoidosis at rest, and in up to 43% on exertion. 6 It is most commonly related to advanced lung disease with fibrosis and bullae198 but about 30% of cases of pulmonary hypertension are not associated with radiographic evidence of lung fibrosis (Fig. 11.31). 199 When pulmonary hypertension is not associated with lung fibrosis, it may respond to corticosteroid therapy.199. and 200.
Occasionally, granulomatous involvement of large or small pulmonary veins causes venous obliteration and postcapillary pulmonary arterial hypertension.201. and 202. Imaging features may include interlobular septal thickening (and resemble those of venoocclusive disease). 202 Rarely, intraluminal filling defects may be seen in the pulmonary veins in patients with sarcoidosis. 201
Systemic vein involvement
Although mediastinal adenopathy is often massive, it rarely causes compression of great veins with subsequent superior vena cava syndrome, a fact that has been ascribed to the lack of perinodal fibrosis in sarcoidosis. 113 A handful of cases of superior vena caval obstruction are described. 203 A single case of subclavian vein obstruction204and of innominate vein obstruction which produced a large exudative pleural effusion is recorded. 205 The rarity of superior vena cava syndrome with sarcoidosis is such that other causes of obstruction should be sought. 206
Pleural sarcoidosis
In sarcoidosis, granulomas may be found on both the visceral and parietal pleura. These pleural granulomas commonly cause no signs or symptoms. 207 On rare occasions, however, they may be associated with a dry pleurisy208 or a pleural effusion that is usually painless209 but may be painful. 210 These effusions are classically lymphocyte-rich exudates that in nearly one-fifth of cases are sanguineous or serosanguineous. 211 Eosinophilic effusions are described. 212
In 1870 cases of sarcoidosis taken from major series in the world literature, 55 had pleural effusions, giving a prevalence of 1.9%.15.33.90.92.93.148.213. and 214. Effusions may be present initially, but more often they develop during the course of established disease, a few months to 16 years after onset. 213 Sarcoid effusions are usually seen in the context of extensive pulmonary disease214 or multisystem involvement. 213 About one-third of the effusions are bilateral, 213 and while they are usually small, large effusions have been described.211.215. and 216. The effusions resolve spontaneously in weeks or months and are almost invariably gone by 6 months. 21 Resolution is usually complete, but about 20% of patients are left with residual pleural thickening.90. and 214. In a CT-based study of 61 patients, pleural effusions were found in 9%, with pleural thickening in 30%. 217
In general, pleural effusion is a relatively minor and incidental finding in the course of sarcoidosis. However, because it is an atypical feature, it may prove a diagnostic puzzle, and other entities such as tuberculosis218 or chylothorax53 must be excluded. 219 Sarcoidosis is associated with a 2–3% prevalence of pneumothorax in several series, usually associated with advanced fibrocystic disease. 220 Uncommonly pneumothorax can be a presenting manifestation of sarcoidosis, associated with multiple pleural granulomata at thoracotomy or subpleural cavitary nodules and bullae at HRCT. 221 Bilateral pneumothorax is also described. 222
Mycetoma
Mycetoma formation (Fig. 11.32) is a well-recognized complication of stage IV cystic sarcoidosis.223. and 224. Indeed, with the declining incidence of tuberculosis, sarcoid is probably now the commonest cause of mycetoma. This is particularly so in the USA, where the proportion of African Americans with sarcoidosis results in more cases of gross fibrobullous disease. 225 In a series of mycetomas reported from Philadelphia, 45% were in patients with sarcoidosis, 226 whereas in a series of 26 patients in the UK only 15% were in patients with sarcoidosis. 227 The frequency of mycetoma in sarcoidosis varies from series to series and ranges between 1% and 10% depending on racial mix of the patients and method of detection.33.90.148.224. and 228. In the subset of sarcoid patients with stage IV fibrocystic disease, the proportion of patients harboring a mycetoma can be as high as 50%.90. and 224.
The imaging features of mycetoma are discussed in Chapter 5. In sarcoidosis, mycetoma formation should be considered when new masslike lesions or pleural thickening develop in patients with stage IV sarcoidosis. 226 Mycetomas are confined virtually to the upper lobes and are commonly bilateral (Fig. 11.32), in 50% of cases in one series. 228 Serum precipitins against Aspergillus spp. are almost always strongly positive. CT is the most sensitive imaging technique.227. and 229.
Role of CT in sarcoidosis
The role of CT in patients with known or suspected pulmonary sarcoidosis remains undefined. Recognition of the characteristic CT pattern of sarcoidosis may help to suggest the diagnosis, and may in some cases be sufficiently specific to obviate the need for biopsy. If a biopsy is desired, CT may provide a ‘road-map’, identifying high-yield areas for transbronchial biopsy. In patients with unilateral adenopathy, in whom there is concern for the presence of lymphoma or other abnormality, CT may help by identifying bilateral nodal enlargement, characteristic nodal calcification, and parenchymal abnormalities suggestive of sarcoidosis. However, CT adds little to the diagnostic evaluation of patients with typical clinical and chest radiographic findings of sarcoidosis.
A study by Mana et al. 230 of 100 patients with sarcoidosis found that CT scans added diagnostically useful information in only one of the 35 patients in whom they were performed. These authors concluded that CT may be helpful in patients with atypical chest radiographic findings, normal chest radiographs, hemoptysis, or suspicion of a second complicating disease, and in those who are lung transplant candidates. To these indications we would add suspected large or small airway disease, and those in whom a diagnosis is not made by transbronchial biopsy. Costabel et al. 36 suggested the following indications for CT: atypical clinical and/or chest radiographic findings; high clinical suspicion of sarcoidosis without typical imaging findings; and suspected complications of lung disease including bronchiectasis, aspergilloma, traction emphysema, infection, or malignancy.
Follow-up CTs have been used to look at the change with time in opacities identified on CT,86.88.118.132.142.144. and 147. giving further insight into the nature of the underlying pathology. Results are not entirely consistent but in general show that nodules, consolidation, ground-glass opacity, and septal lines are potentially reversible (see Figs 11.17 and 11.20) while irregular coarse lines, cysts, honeycombing, traction bronchiectasis, and distortion are permanent. However, a considerable proportion of ‘potentially reversible’ opacities do not improve with treatment; for example 57% and 39% of nodules did not change in two follow-up studies,118. and 142. and ground-glass abnormality and consolidation evolved to a fibrotic pattern on long-term follow-up in another study. 132
Identification of the extent and predominant pattern of abnormality on CT may be helpful in predicting the degree of physiologic impairment. Patients with focal alveolar abnormality or masses tend to have little or no physiologic impairment, while those with profuse nodules or architectural distortion tend to be significantly impaired. 161 As mentioned above, patients with air-trapping on expiratory imaging will usually have a component of airflow obstruction. However, the overall level of correlation between extent of CT findings and physiologic abnormality is relatively weak.
Role of other imaging modalities
Gallium-67 is commonly taken up in sarcoid lesions; therefore, gallium-67 scintigraphy was previously used to assist in diagnosis of sarcoid, and in determination of disease ‘activity’. Characteristic scintigraphic patterns include uptake in bilateral mediastinal nodes, producing the ‘lambda’ sign, and symmetric uptake in salivary and lacrimal glands, producing the ‘Panda’ sign. 231 However, the technique is now rarely used because of lack of specificity of the pulmonary findings, 232 and because of the greater sensitivity of [18 F]2-fluoro-2-deoxy-D-glucose (FDG) positron emission tomography (PET)-CT scanning for detection of organ involvement, particularly extrathoracic involvement.233. and 234.
PET scans with FDG may show increased metabolism in affected lung parenchyma235 and nodes236 (Fig. 11.33). In a study of 137 patients, evaluated with FDG-PET for cardiac involvement by sarcoidosis (n = 88) or for other reasons, extracardiac uptake was identified in 139 of 188 scans performed. 237 Mediastinal nodal uptake was the most common abnormality, identified in 54, extrathoracic nodal uptake in 30, and lung uptake in 24. In 20 patients (15%), FDG-PET scan findings indicated the presence of abnormality that was not detected by physical examination, chest radiograph, or CT, including uptake in the lung, mediastinal lymph nodes, peripheral lymph nodes, skin, muscle, and bone. In these 20 patients, the PET scan findings led to a diagnostic biopsy at different sites in five patients. Pulmonary uptake was visible in four of 14 patients with radiographic stage I disease, in 16 of 22 patients with stage II or III disease, but, interestingly, in only three of 22 patients with fibrotic, stage IV disease. On repeat scanning, 11 steroid-treated patients showed decreased standardized uptake values (SUVs) in the affected areas, but the paper does not indicate how many patients were unchanged. These findings suggest that FDG-PET may occasionally have a role in identifying appropriate biopsy sites, or in determining whether patients with fibrotic abnormality have residual active disease. Other studies have shown that FDG-PET commonly identifies occult bony or soft tissue sites of sarcoidosis, 238 and can also identify cardiac involvement. 239 Sequential scans may be helpful in monitoring response to treatment, particularly in atypical, complex, and multisystemic disease. 233 Kaira et al. 798 studied 24 patients with sarcoidosis and suspected malignancy using fluorine-18-alpha-methyltyrosine (18-FMT) and found that the 18-FMT scans were always negative, allowing distinction from a control group of patients with lung cancer, in whom the 18-FMT scans were abnormal in 88%.
MRI of the lung or mediastinum does not have a role in evaluation of sarcoidosis. However, magnetic resonance (MR) with delayed hyperenhancement has a well-established role in the diagnosis of cardiac involvement by sarcoidosis. 240
EOSINOPHILIC LUNG DISEASE
Despite an explosion of information about the eosinophil, the function of this cell remains unclear. However, it is seen in a variety of inflammatory processes, and a group of pulmonary disorders is associated with the presence of eosinophils (Table 11.1). The term pulmonary eosinophilia was introduced in 1952 to describe a group of diseases ‘in which pulmonary infiltration on the radiograph is accompanied by blood eosinophilia but in which pneumonia, hydatid disease of the lung, Hodgkin’s disease, and sarcoidosis can be excluded’. 242 The 16 patients in this study were classified into five groups: simple pulmonary eosinophilia (Löffler syndrome), prolonged pulmonary eosinophilia, tropical eosinophilia, pulmonary eosinophilia with asthma, and polyarteritis nodosa. Later authors have widened this concept, 243 using the term eosinophilic lung disease to include all disorders associated with blood and/or tissue eosinophilia which affect major airways and/or lung parenchyma. Blood eosinophilia is generally taken to mean an eosinophil count of more than 0.4 × 109/ L, though some set the level at 0.5 × 109/ L. It is important to note that it is not necessary to have a blood eosinophilia to make a diagnosis of eosinophilic lung disease, and indeed BAL has become widely used for more sensitive and specific diagnosis of eosinophilic pneumonia. In eosinophilic pneumonias, the percentage of eosinophils in BAL fluid is usually greater than 10% (with normal being less than 1%). 241 The term pulmonary infiltration with eosinophilia (PIE) is synonymous with the term pulmonary eosinophilia.
| Disease | Etiology | Blood eosinophilia | Length of history | Radiologic features |
|---|---|---|---|---|
| Eosinophilic pneumonia of unknown cause | ||||
| Acute eosinophilic pneumonia (EP) | Idiopathic | Uncommon at presentation | 1–5 days | Diffuse, or basal ground-glass/reticular opacities; effusions |
| Löffler syndrome (simple pulmonary eosinophilia) | May be idiopathic; often due to ABPA, drugs, or parasitic infection | Usual | Fleeting airspace opacities | |
| Chronic EP | Idiopathic | 95% > 1× 109/L | 2 weeks to 6 months | Dense, peripheral consolidation, often upper lobe predominant; may be migratory |
| Hypereosinophilic syndrome | Idiopathic | Marked (>1.5× 109/L) | ≥6 months | Heart failure with edema; effusions; rare parenchymal opacities |
| Churg–Strauss syndrome | Usually idiopathic vasculitis | Usual | Weeks to months | Consolidation (may be migratory), nodules, cavities, bronchiectasis |
| Eosinophilic bronchitis | Unknown | Not present | >8 weeks | Airway wall thickening |
| Eosinophilic pneumonia of known cause | ||||
| Allergic bronchopulmonary aspergillosis (ABPA) | Immune response to Aspergillus | Usual in acute ABPA | Days to weeks | Migratory opacities; mucoid impaction; central bronchiectasis |
| Bronchocentric granulomatosis | May be related to Aspergillus | Common | Mass lesions, alveolar opacities | |
| Drug reactions | Nonsteroidal anti-inflammatory drugs; antibiotics241 | Usually | Days to weeks | Ground-glass/reticular |
| Tropical eosinophilia | Lymphatic filarial parasites; Wuchereria bancrofti and Brugia malayi | 3000–80 000/mm3 | Variable | Reticulonodular basal opacities; migratory opacities; chest radiograph may be normal |
| Parasitic infection | Many parasites (Dirofilaria, Ascaris, Strongyloides, Toxocara) | Usual | Variable | Migratory opacities; chest radiograph may be normal |
The possible causes of pulmonary eosinophilia in a patient under investigation can usually be markedly reduced by taking into account a few key historical, clinical, and laboratory findings; notably, work exposure, ethnic background, travel in endemic areas, and a history of asthma, atopy, and any medication. Useful information from first-line investigations includes the magnitude of blood eosinophilia, total serum IgE, the presence of skin sensitivity, serum Aspergillus precipitins, and cysts, ova, or parasites in the stool.
Eosinophilic pneumonias
The eosinophilic pneumonias are classified on the basis of their severity and chronicity (Table 11.1). 244 However, it should be noted that substantial overlap exists among these entities, and specific categorization may sometimes be difficult. 245 Simple eosinophilic pneumonia (SEP) (Löffler syndrome) has classically been distinguished from chronic eosinophilic pneumonia (CEP), on the basis of duration of symptoms. 242 SEP is defined as lasting less than 1 month, while CEP lasts more than 1 month. The 1-month dividing line is rather arbitrary and not universally applied, so that the distinction between the two types is not always clear, and this is particularly true now that the natural history is commonly modified by steroids. Nevertheless the division is useful clinically, as the majority of chronic, cryptogenic eosinophilic pneumonias (CEP) form a relatively homogeneous group. The more recently recognized acute eosinophilic pneumonia (AEP)246. and 247. differs from SEP in that patients are highly symptomatic and in respiratory failure.
Acute eosinophilic pneumonia
AEP was first described in 1989246. and 247. and is considered to represent a distinct clinical entity with the following diagnostic criteria: (1) acute febrile illness of less than 5 days’ duration, (2) hypoxemic respiratory failure, (3) alveolar or alveolar/interstitial chest radiographic opacity, (4) bronchoalveolar lavage eosinophil level of more than 25%, (5) prompt and complete response to steroids without relapse on withdrawal, and (6) absence of parasitic, fungal, and other infection. It is characterized histologically by acute and organizing diffuse alveolar damage, with eosinophils present mainly in the pulmonary interstitium. 248
AEP commonly occurs in patients who have recently begun to smoke cigarettes. In a series of 33 cases from Japan, 249 21 had just begun to smoke, two had restarted smoking, and six had substantially increased their cigarette consumption. The median interval between onset of smoking and onset of AEP was usually less than 1 month. 249 In a series of 22 patients from France, six of eight cigarette smokers had begun to smoke 1 week to 3 months before the onset of AEP. 250 There may also be a relationship to inhalation of other types of materials such as gasoline fumes250 and dusts. Eighteen cases were identified among military personnel deployed in Iraq in 2003/2004, and evaluation of these cases suggested a relationship to recent onset smoking, and to exposure to fine dust. 251 AEP, presumed to be related to dust exposure, was also described in a firefighter who had spent 13 days working at the World Trade Center site immediately after September 11, 2001. 252 AEP may also be related to drugs including minocycline, heroin, and progesterone. 253
The average age at presentation is about 30 years and there is no sex predilection. 254 Principal symptoms are those of cough, dyspnea, fever, and chest pain coming on over a period of a few days. On examination patients are tachypneic and hypoxic. A history of asthma or atopy may or may not be present.255. and 256. Respiratory failure, with requirement for mechanical ventilation, develops in most patients. 250 There is bilateral chest radiographic opacity and the usual presumptive diagnosis is of a fulminant chest infection or acute respiratory distress syndrome (ARDS). Blood eosinophil levels are elevated in about 35% of cases at presentation, but become abnormal at some point in the course of disease in 70%. 250 The key to diagnosis, however, is provided by bronchoalveolar lavage showing a greatly raised eosinophil percentage which may be in the order of 30–80%.250. and 254. The other important diagnostic clue lies with chest imaging, as detailed below. Acute eosinophilic pneumonia responds satisfactorily to steroids and patients become symptom-free with essentially normal respiratory function. The condition does not relapse. 254
AEP is one of the few forms of acute interstitial pneumonia in which the diagnosis may be strongly suspected based on chest radiographic and CT features (Box 11.7). As stated above, radiographic findings (Fig. 11.34)246.254.256.257.258. and 259. are usually a mixture of airspace and interstitial opacity, including septal lines.254. and 260. These changes are bilateral but range from being diffuse to localized. Either consolidation or septal thickening may be found in isolation. 254 About 70% of patients have pleural effusions (bilateral more commonly than unilateral) at presentation,261. and 262. and during the course of the illness essentially all patients develop bilateral effusions. 259 In a series of 22 patients studied by Philit et al., 250 pleural effusions were visible on the initial chest radiograph in only two patients, but were seen on CT in 10 of the 14 patients who underwent CT. This suggests that the pleural effusions were not visualized in some patients on chest radiograph, either because of the associated parenchymal abnormality or because erect frontal and lateral radiographs were not obtained in these ill patients.
Box 11.7
• Pleural effusions
• Septal thickening
• Peribronchovascular thickening
• Ground-glass abnormality
• Consolidation
CT may be very helpful in suggesting the diagnosis, with typical findings of septal lines, pleural effusions, and areas of consolidative and ground-glass opacity (Fig. 11.34).257. and 259. Areas of ground-glass abnormality and/or consolidation are found in almost all patients.261.262. and 263. Thickened interlobular septa are seen in about 75%, with thickening of bronchovascular structures in about the same number, and pleural effusions in 70–80%. Pleural effusions may be small or large. About 30% of cases have an upper lung predominance of abnormality. 262 The distribution of consolidation and ground-glass abnormality is peripheral in more than 50% of cases, but may be central or random. 263
In patients with acute respiratory failure, the demonstration by chest radiograph or CT of prominent interlobular septal thickening, thickening of bronchovascular bundles, or pleural effusions should prompt the recommendation of BAL to identify the presence of eosinophilia, and make the diagnosis of AEP. If eosinophilia is demonstrated, lung biopsy is probably not indicated. 264 This is an important diagnosis to make, because survival with appropriate management is close to 100%, 250 much better than in most other forms of acute respiratory failure.
Simple eosinophilic pneumonia (Löffler syndrome)
The characteristic features of simple pulmonary eosinophilia or Löffler syndrome are (1) blood eosinophilia, (2) absent or mild symptoms and signs (cough, fever, dyspnea), (3) one or more nonsegmental pulmonary consolidations that are transitory and/or migratory, and (4) spontaneous clearing of consolidations. Originally, opacities were described as disappearing within 6–12 days, 265 but this interval is now generally extended to a month. 242 Presentation has been noted to vary seasonally. 266 The prognosis is excellent. 266
Löffler syndrome may be idiopathic (cryptogenic), or it may result from a variety of inciting agents, particularly allergic bronchopulmonary mycosis, drugs, parasites, and miscellaneous agents such as nickel carbonyl. 267 It seems likely that some, if not all, of Löffler’s original cases were related to ascariasis. 266 In developed countries, the commonest cause of Löffler syndrome is probably allergic bronchopulmonary aspergillosis (see Fig. 11.46 below). 241 Careful evaluation will lead to an identification of a cause in most cases of Löffler syndrome, and, indeed, there is some doubt as to whether this syndrome is ever truly idiopathic. 241
Pathologically, there is an eosinophilic pneumonia with edema and an eosinophilic infiltrate in both alveoli and interstitium. Radiographically the findings are one or more fairly homogeneous, nonsegmental consolidations that can be small or so large as to occupy much of a lobe. They are transitory and may be migratory, disappearing from one area and appearing in another. They have a tendency to be peripherally located. Pleural effusions, mediastinal adenopathy, and cavitation are not described.
Recently, it has been asserted that pulmonary eosinophilia accounts for a substantial proportion of transient ground-glass, part-solid, or solid nodules incidentally identified when chest CT is used in screening programs for lung cancer. 268 In a review of 40 such cases, about 50% had solitary opacities, and 50% had multiple lesions. 269 The opacities were usually either solid nodules with ground-glass halos or poorly defined solid nodules. Nodules were relatively large, with a mean diameter of 1.6 cm. They were usually distributed in the lower zones and peripherally in the lung, and usually resolved completely on follow-up. It is possible that some of these cases of transient eosinophilic pneumonia may have been due to subclinical parasitic infection.
Chronic eosinophilic pneumonia
CEP is a cryptogenic form of eosinophilic lung disease with consistent and characteristic clinicopathologic features. Pathologically, there is an eosinophil-rich exudate in alveoli and interstitium.270. and 271. Angiitis is mild, fibrosis sparse, and necrosis very rare. 270 Many patients present in middle life but prevalence remains high from the third to seventh decade, 272 range 7–77 years; women outnumber men by 2 : 1; 271 50% of patients are atopic, 40% asthmatic, and 5–10% have allergic rhinitis and nasal polyps. 272 Asthma can antedate the condition by many years273 or can develop with the onset of CEP. The symptoms are usually highly characteristic, range from mild to severe, and have often been present for several months. Typical symptoms are cough with mucoid sputum; high fever, particularly in the evenings, often accompanied by drenching night sweats; dyspnea; wheeze; malaise; and marked weight loss (8–12 kg). Occasionally there is chest pain and hemoptysis. Blood eosinophilia is common, but not universal, occurring in nearly 90% of patients, 272 ranging from mild to marked. 271 There is sputum eosinophilia in less than 50% of patients. 272 Total white blood cell count and the erythrocyte sedimentation rate (ESR) are raised. Serum IgE is normal or only minimally elevated, 271 allowing distinction from those conditions such as allergic bronchopulmonary aspergillosis and tropical and parasitic pulmonary eosinophilias in which serum IgE levels are markedly elevated. However, there have been a number of reports of markedly raised IgE levels. 274 BAL demonstrates high eosinophil percentages – in the order of 25% or more. The principal pathologic findings are filling of alveoli with eosinophils and macrophages, sometimes with necrosis and the formation of eosinophilic abscesses. Foci of organizing pneumonia may be seen. Changes are not confined to alveolar spaces and there is typically an interstitial pneumonia as well, but fibrosis is not usually seen. 241
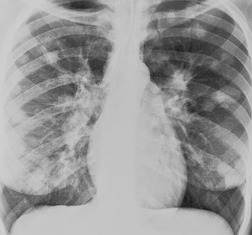
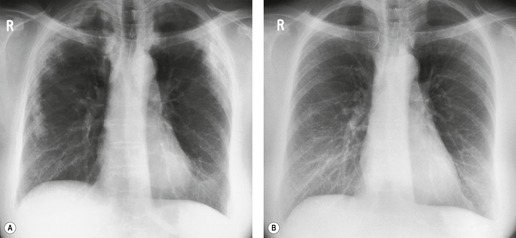
Some patients with chronic eosinophilic pneumonia have an imaging pattern that is virtually pathognomonic (Box 11.8). At its most classic, the pattern, seen in two-thirds of patients, 272 consists of peripheral, nonsegmental, homogeneous consolidations sometimes with an air bronchogram, the so-called ‘photographic negative of pulmonary edema’ (Fig. 11.35). 275 These opacities lie against the chest wall and may surround the lung or just occupy one or two zones, particularly the apices (Fig. 11.36). In one series the zonal distribution was 46% upper, 40% mid, and 14% lower. 276 About 50% of patients have bilateral consolidation which may cloak the upper and outer aspects of the lung in a very characteristic fashion (Fig. 11.35).242. and 277. However, this characteristic peripheral distribution is seen in only a minority of cases (Fig. 11.37).244. and 272. The consolidation may even be predominantly perihilar in distribution. 276 A common pattern is mixed peripheral and central consolidation (Fig. 11.37). 276 Opacities may appear in one lung to be followed by others on the opposite side, or they may disappear spontaneously. If they resolve they usually recur in the same place.276. and 277. Unilateral diffuse consolidation may occur. 278 Isolated lesions in the upper zone may closely mimic those of tuberculosis. Other features that are infrequently seen include pleural effusions (2%), cavitation (5%), 272 and occasional mediastinal lymphadenopathy, best appreciated on CT.279.280. and 281.
Box 11.8
• Consolidation
• Ground-glass abnormality
• Peripheral predominance
• Upper lung predominance
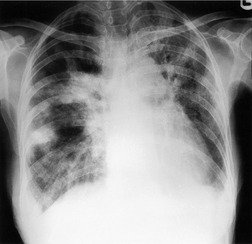 |
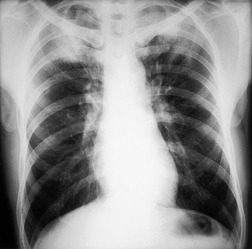
| Fig. 11.37 Chronic eosinophilic pneumonia (CEP). Radiograph shows multifocal peripheral and central consolidation. This is a recognized pattern but less common than the one shown in Fig. 11.35. Both costophrenic angles are blunted in this patient. Pleural effusions are very unusual in CEP. |
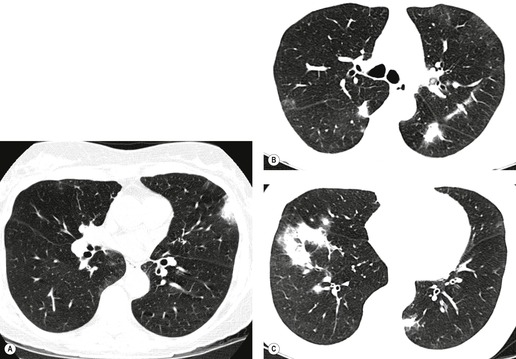
CT is often unnecessary to confirm the diagnosis of CEP, if typical imaging findings are associated with blood eosinophilia. In general, CT studies confirm the chest radiographic findings, showing strikingly peripheral, multifocal consolidation or ground-glass abnormality (Fig. 11.38A), though there may also be areas of nonperipheral consolidation.280. and 282. In one study, ground-glass opacity was as common a finding as consolidation. 282 However, it was infrequently the only finding (in 12% of patients) and was usually close to the margins of consolidated areas. 282 The bilateral subpleural distribution is universal in patients who are scanned within 1 month of the onset of symptoms. 282 Patients with CEP who are imaged more than 1 month after onset of symptoms have a different pattern on CT. The consolidation tends to be more patchy (Fig. 11.38B,C), and though the opacities remain peripheral, the subpleural zone is often relatively clear. A characteristic feature in these more chronic cases is the presence of a dense bandlike structure parallel and about 1–2 cm deep to the chest wall (Fig. 11.38B), which may traverse the fissures.279. and 282. In chronic cases of CEP, dense fibrosis and lung distortion may rarely be seen.
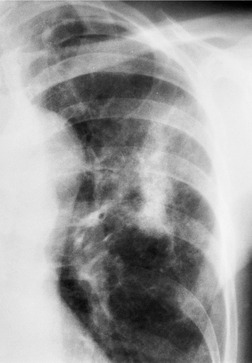
The disease occasionally remits spontaneously; 277 however, treatment is usually required, and CEP is remarkably sensitive to steroid therapy (Fig. 11.35). Rapid clearing is usually seen within a few days, with complete clearing by 1 month. 270 In fact some authors recommend a therapeutic trial of steroids in previously well patients with classical clinical and imaging features. 272 Radiographic resolution is usually complete. The characteristic bandlike opacities parallel to the chest wall may be seen during the resolving phase (Fig. 11.39).272.277. and 282. Clinical relapse occurs in about 30–60%, 283 and in a review of 62 patients 21% relapsed during reduction of steroid dose and 58% after discontinuation. 272 The majority of patients need long-term low-dose steroids284 and a proportion develop late-onset asthma. 277 In some instances, patients who initially have all the features of CEP go on to develop the Churg–Strauss syndrome285 or a diffuse vasculitis.277. and 286.
The imaging features of CEP overlap with those of cryptogenic organizing pneumonia, but may be distinguished by recognition of blood eosinophilia. CEP must also be distinguished from Churg–Strauss syndrome, which should be suspected if there is evidence of cardiac or other systemic involvement. 244
Allergic bronchopulmonary mycosis
Allergic bronchopulmonary mycosis is almost certainly the most common cause of eosinophilic lung disease in developed countries. Allergic bronchopulmonary aspergillosis (ABPA) accounted for 78% of the patients with a diagnosis of pulmonary eosinophilia in a series of 143 in the UK who were admitted to a tertiary referral center. 287 First described in 1952, 288 the disease is characterized by asthma, radiographic pulmonary opacities, blood eosinophilia, and evidence of allergy to antigens of Aspergillus species. Its importance lies in the fact that recurrent acute episodes cause progressive bronchial damage that can be controlled by steroid administration. 289
Though it may be due to a wide variety of organisms, Aspergillus species are by far the most common causative agent, resulting in ABPA. In more than 90% of patients, the species involved is Aspergillus fumigatus, but occasionally other species are implicated, including Aspergillus flavus, Aspergillus niger, Aspergillus nidulans, 290Aspergillus terreus, 291Aspergillus oryzae, 292 and Aspergillus ochraceus. 293 In addition there are isolated case reports of an ABPA-like syndrome caused by fungi other than Aspergillus spp., including Candida albicans, Curvularia (see Fig. 11.41 below) and at least seven other genera.294. and 295. These conditions may be more difficult to diagnose because Aspergillus precipitins may be absent.
In ABPA, a hypersensitivity reaction develops to Aspergillus spp. that grow as a mycelial plug in proximal airways, usually the second or third order bronchi. 296 Tissue invasion is either absent290 or very limited. 297 The factors that favor the initial airway colonization by Aspergillus are unclear, 290 but in part are probably related to the almost universal presence of asthma and atopy in affected patients. Other important factors are the size of the spores (2.5–3.0 µm), which favors inhalation and airway deposition, and their thermotolerance, which allows hyphal growth at body temperatures.294. and 298. Certainly, once the fungus gets a foothold, local damage will promote further colonization.
The immunology and pathogenesis of ABPA is complex. CD4+Th-2 lymphocyte activation is thought to be important in releasing mediators that increase eosinophil production, and stimulate B cells to produce IgE and IgG production. 299 Because antigen production is localized to the mycelial plug in the proximal airway, tissue damage tends to be greatest in this region, giving rise to granulomatous airway inflammation that results in the characteristic proximal bronchiectasis. In addition, however, there appears to be a separate pathologic process centered on the lung parenchyma, giving an eosinophilic pneumonia which does not produce permanent damage. 294
Typically, ABPA occurs in patients with atopy and longstanding asthma. A few patients, particularly older ones, develop asthma concurrently with their first attack of ABPA. 287 Although unusual, the disease is well-recognized in nonasthmatic patients.287. and 300. Overall there is a slight female preponderance.287.301. and 302. It may occur at any age,302.303. and 304. but typically presents between 20 and 40 years of age. The entity is familial in about 5% of cases. 305 There is an association with several HLA alleles. 306 The length of the interval between the onset of asthma and ABPA is inversely related to the age of onset of asthma. McCarthy and co-workers307 found that with asthma beginning before 10 years of age, there was an average gap of 24 years, but with late-onset asthma (30 years of age or more), the mean gap was only 3.5 years. Late-onset asthma was associated with more frequent attacks of ABPA and greater lung damage. 307 ABPA also occurs in patients with cystic fibrosis in whom there is an approximately 10% prevalence. 266
In about 10% of patients, allergic bronchopulmonary mycosis may be associated with allergic fungal sinusitis. 308 This condition is characterized by accumulation of mucin in the sinuses, associated with polyposis in the sinuses and nose. It is associated with a wider range of organisms than allergic bronchopulmonary mycosis, including Curvularia and Bipolaris spp.309.310.311. and 312. On imaging, it is characterized by mucosal thickening and hyperattenuating sinus contents, often associated with bony erosions.313. and 314.
ABPA runs a relapsing and remitting course and an acute attack may be indistinguishable from an uncomplicated exacerbation of asthma. 294 Patterson et al. 315 described five clinical stages of ABPA: acute, remission, exacerbation, steroid-dependent asthma, and fibrosis. These must not be regarded as phases of disease, as patients need not pass from one stage to the next in orderly progression. Characteristic symptoms in the acute stage are wheeze, dyspnea, and a cough that is often productive and associated with minor hemoptysis. Systemic symptoms such as fever, malaise, and weight loss are common. About 50% of patients have pleuritic pain, and about the same percentage give a history of coughing up sputum plugs. 287 These contain fungal mycelia and are important pointers to the diagnosis. 287 Plugs are about l–2 cm long, firm, friable, and pelletlike. 287 An abnormal chest radiograph, blood eosinophilia, and an immediate skin ‘wheal and flare’ reaction to Aspergillus antigens are characteristic of the acute phase. Eosinophilia is usually mild to moderate, with 74% of patients having counts between l × 109/L and 3 × 109/L. 287 The eosinophil level may be depressed by steroid treatment. Serum precipitins against Aspergillus antigens are detected in about 90% of patients in the acute phase, particularly if the serum is concentrated.287.294.301. and 316. It must be remembered, however, that this test is nonspecific in that 12–25% of patients with extrinsic asthma will have a positive precipitin test. 317 Both nonspecific and Aspergillus antigen-specific IgE are greatly raised, perhaps 20 times normal. 316 Smaller elevations may be found in simple asthma. As with the finding of positive precipitins, elevation of IgE per se is not diagnostic of ABPA. Nevertheless the IgE level is probably the single most useful laboratory test in ABPA since levels correlate with disease activity and a normal level virtually excludes the diagnosis.266. and 316. Criteria for the diagnosis of ABPA have changed over time as knowledge about the disease has become more sophisticated; yet even now there are no universally accepted diagnostic criteria. Rosenberg and co-workers316 have set out the following major and minor criteria and suggest that if the first six of the major criteria are satisfied, the diagnosis of ABPA can be made with reasonable certainty. The major criteria for ABPA are:
1. Asthma
2. Blood eosinophilia
3. Immediate skin reactivity to Aspergillus antigen
4. Precipitin antibodies to Aspergillus antigen
5. Raised serum IgE (nonspecific/specific)
6. History of radiographic pulmonary opacities
7. Central bronchiectasis.
The minor criteria for ABPA are:
1. A. fumigatus in sputum
2. History of expectorating brown plugs
3. Late skin reactivity to Aspergillus antigen.
ABPA can be present without bronchiectasis,266.318. and 319. and ABPA without bronchiectasis appears to carry a better prognosis, with fewer exacerbations.320. and 321. In one study, 11 of 31 patients with serologically diagnosed ABPA did not have bronchiectasis; 321 these patients had less severe disease than those with bronchiectasis, and did not subsequently develop bronchiectasis on follow-up over 2 years. 322 The prevalence of bronchiectasis may decrease with increasing use of serology to screen for ABPA: in a more recent study of 155 patients with ABPA, 37 (24%) had a normal findings on CT. 323
In a prospective study of 255 patients with asthma, Eaton et al. 324 found that 47 patients had positive skin prick tests for A. fumigatus, of whom 35 underwent CT. Central bronchiectasis was found on CT in 12 of these patients, eight of whom met all of the criteria for ABPA. These investigators found that performing CT in patients with skin prick positivity who had histories of sputum plugs, eczema, or steroid dependency would identify all cases of ABPA with bronchiectasis. These authors proposed the following minimal criteria for ABPA: asthma, skin prick test positivity, and central bronchiectasis.
A great variety of imaging changes are found in ABPA, and may be related to the phase of the disease.301.302.304.307.325.326. and 327. The imaging findings are best considered as acute and transient or chronic and permanent (Table 11.2). 307
| Major | Minor |
|---|---|
| Acute (transient) | |
| Consolidation (80%) | Airway wall thickening |
| Mucoid impaction (bronchocele) (30%) | Small nodules |
| Atelectasis (20%) | Pleural effusions |
| Chronic (permanent) | |
| Bronchiectasis | Pleural thickening |
| Mid/upper zone volume loss and scarring | Mycetoma |
| Small nodules | |
| Linear scars | |
The transient changes are listed in Table 11.2. Consolidation ranges from patchy subsegmental or smaller opacities (Fig. 11.40) to complete lobar or multisegmental opacity (Fig. 11.41). Areas of consolidation show little if any zonal predilection and are often multiple. The segmental configuration contrasts with the nonsegmental consolidation seen in other eosinophilic states. The pathologic basis of the consolidation is unclear: it might be related to airway obstruction or eosinophilia. Although consolidation is usually transient, it can last for 6 weeks or more. Phelan and Kerr302 described some patients with apparently permanent consolidation, perhaps due to endobronchial obstruction. When consolidation clears, it often leaves residual bronchiectasis (Fig. 11.41). Consolidation may recur in 25–50% of cases, often in the same area. Radiographic consolidation may be asymptomatic in 20–30% of patients,289. and 304. and ABPA is one of those conditions in which gross radiographic changes may be accompanied by little in the way of symptoms. 307
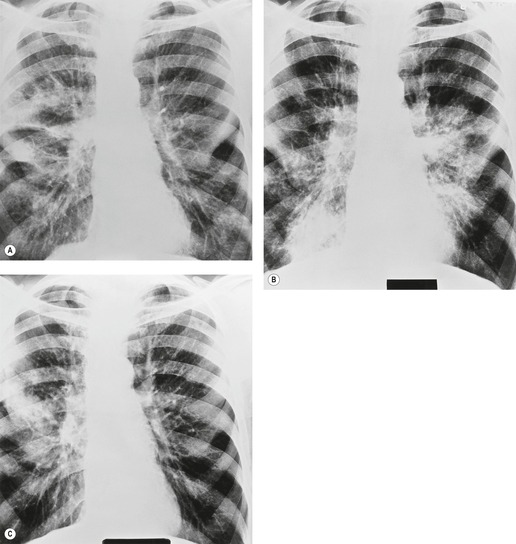
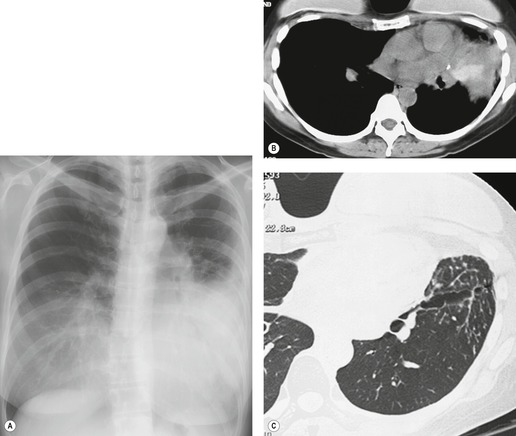
The most common transient change is mucoid impaction (bronchocele). In this condition, an airway becomes obstructed and distended by retained secretions, but the subtended lung remains aerated by collateral air drift, allowing direct visualization of the impacted airway. Mucoid impaction is characterized by a sharply demarcated, bandlike opacity, sometimes branching (Fig. 11.42), which points to the hilum, usually about 2–3 cm long and some 5–8 mm wide. Rounded opacities are produced by linear bronchoceles seen end-on. Impacted airways are usually proximal. They are often inseparable from the hilum and may simulate lymphadenopathy327 or a mass. Another variant is produced by distal bronchiectatic airways which, when impacted, give a band shadow with a club-shaped end – the ‘gloved finger shadow’. Bronchoceles show a strong upper zone predilection (Figs 11.42 and 11.43). 304 They disappear once their contents have been coughed up, leaving dilated bronchi, manifested as ring or parallel line opacities (Fig. 11.43). Like consolidation in ABPA plugging may recur at the same site. Occasionally they persist for many months. On CT, the bronchoceles are seen as bandlike abnormalities in the expected position of the bronchi (Fig. 11.42). Their attenuation may be similar to that of water or soft tissue, or they may be hyperattenuating (Figs 11.41 and 11.42). 328 The hyperattenuation is presumed to be due to calcium329 or metallic ions within chronically inspissated mucus, and is found in 20–30% of cases.330. and 323. Relapse of ABPA occurred in 23% of one series of 155 patients, and was more common in those with more severe bronchiectasis or with hyperattenuating mucus. 323 Another major transient manifestation of ABPA is atelectasis (Fig. 11.44), which ranges in frequency from 3%304 to 46%. 296 It may be subsegmental, segmental, lobar, or even affect a whole lung.302.326. and 331. Like consolidation and mucoid impaction, collapse has a tendency to recur in the same area. On CT, mucus plugs may be seen within the areas of atelectasis if they are hyperattenuating or of water attenuation. There have been a few reports of pleural effusion.304. and 332.
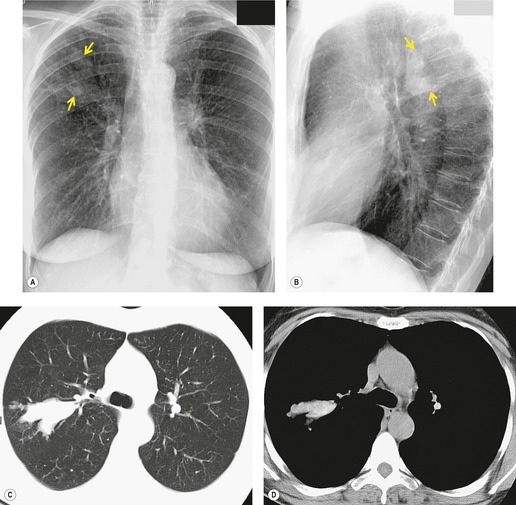
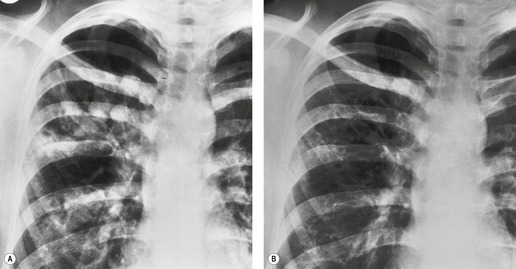
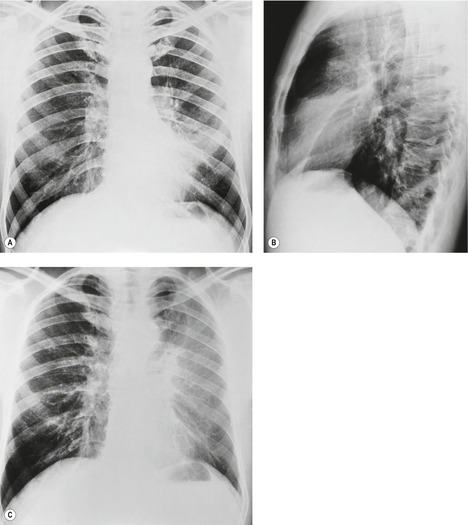
Permanent changes (Table 11.2) are of importance because, first, they indicate irreversible lung damage and, second, they may be the only clue that an asthmatic patient has ABPA when a patient is in remission. Bronchiectasis is responsible for most of the permanent radiographic changes. The characteristic finding (Box 11.9) is proximal bronchiectasis, usually affecting first and second order subsegmental bronchi (Figs 11.45 and 11.46). Beyond the proximal bronchiectasis, more distal airways remain normal and patent, though a tree-in-bud pattern of abnormality is common on CT. Proximal bronchiectasis has been considered highly specific for ABPA316 and in a selected patient population of asthmatic patients, with skin tests positive for A. fumigatus, this is probably so. 333 However, central bronchiectasis may also be found in other conditions such as cartilage deficiency syndromes, primary ciliary dyskinesia, and idiopathic bronchiectasis. 334
Box 11.9
• Involve segmental and subsegmental bronchi
• Varicose or cystic
• Thin-walled
• Associated mucoid impaction (which may be hyperattenuating)
• Associated centrilobular nodularity
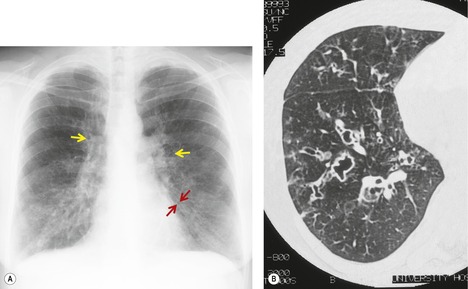
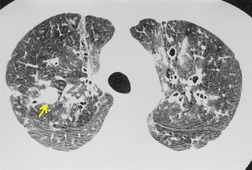
Several papers have compared the CT features of ABPA with those of uncomplicated asthma.335.336. and 337. The prevalence of bronchial dilatation in patients with severe or chronic asthma ranges from 17% to 30%, compared with 90% or more in ABPA.335. and 336. However, the bronchial dilatation in asthma is usually mild, cylindric, and distal, in contrast to the more severe proximal varicose or cystic bronchiectasis of ABPA. 335 Other distinguishing features on CT include a higher prevalence of centrilobular nodules and mucoid impaction in ABPA.335. and 336. The presence of bronchiectasis in three or more lobes is suggestive of ABPA. 336
Parenchymal scarring representing the fibrotic stage of ABPA commonly follows bronchiectasis, and is manifest by linear abnormality and lobar shrinkage (Figs 11.46 and 11.47). Mirroring the distribution of bronchiectasis, these features have a strong upper zone predilection, with 78% being so distributed in one series. 307 Such lobar shrinkage is accompanied by a variety of ring and linear opacities. Lower lobe shrinkage, though described, is very unusual. 302 Although there is often chronic lobar shrinkage, the overall lung volume is frequently increased, reflecting airflow limitation, emphysema, and bulla formation. Between 30% and 40% of patients in one series showed overinflation. 302 Pleural thickening is not a major feature, having a prevalence of 18% in the same series, though pleural thickening was seen on CT in 14 of 17 cases of ABPA in one series. 333
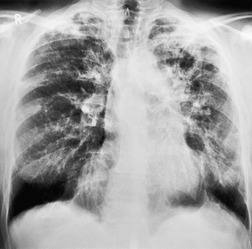
Mycetomas may form in the bronchiectatic cavities. In one series of 111 patients with ABPA, eight had mycetomas, predominantly mid-zonal. 307 Since mid-zone mycetomas are unusual in other conditions, it has been suggested that a mycetoma found in this position should raise the possibility of underlying ABPA. 307 It is of interest that an ABPA-type syndrome has been recorded as developing as a result of a mycetoma lodged in a tuberculous cavity. 338
ABPA in cystic fibrosis
The reported prevalence of ABPA in patients with cystic fibrosis has varied from 1% to 50%, depending on diagnostic criteria and on the aggressiveness of screening.339. and 340. The prevalence of ABPA in cystic fibrosis patients in the USA appears to be about 2%, 339 compared with 7.8% in Europe.340. and 341.Box 11.10 shows the criteria for diagnosis of ABPA in this condition, based on a consensus statement. 340 The diagnosis of ABPA in cystic fibrosis is difficult, and may often be delayed, because many of the diagnostic criteria overlap with common manifestations of the disease. 340
Box 11.10
Classic case
1. Acute or subacute clinical deterioration not attributable to another etiology
2. Serum total IgE concentration of >1000 IU/mL
3. Skin test positivity to Aspergillus
4. Precipitating antibodies or IgG antibody to A. fumigatus
5. New or recent abnormalities on chest radiography (infiltrates or mucus plugging) or chest CT (bronchiectasis) that have not cleared with antibiotics and standard physiotherapy
Minimal diagnostic criteria
• Criteria 1, 2, 3, and either 4 or 5
From an imaging viewpoint, the problem with diagnosis of ABPA in cystic fibrosis is that bronchiectasis and pulmonary opacities are common in uncomplicated cystic fibrosis. The published criteria for diagnosis of ABPA in cystic fibrosis (Box 11.10) suggest that such abnormalities may support the diagnosis of ABPA if they are new and have not responded to standard treatment. The type and distribution of the bronchiectasis may assist in the diagnosis, since central bronchiectasis is relatively uncommon in cystic fibrosis, though common in ABPA. In a study of patients with bronchiectasis, central bronchiectasis without peripheral bronchiectasis was seen in 11 (37%) of 30 patients with ABPA and in only one (7%) of patients with mild cystic fibrosis. 334 Although varicose or cystic bronchiectasis is quite common in cystic fibrosis, it appears to be more common in ABPA. 340 High-attenuation mucus plugs have been reported in ABPA, 330 but not in cystic fibrosis, so these might be suggestive of ABPA in the correct clinical context.
Drug-induced eosinophilic lung disease
A large number of drugs has been recorded as producing pulmonary opacities as part of a hypersensitivity response together with blood eosinophilia (see p 509). Patients generally develop symptoms within days or a few weeks of starting the drug and some develop an associated rash and pyrexia, which provide helpful clues as to the nature of the imaging opacity. A variety of chest radiographic patterns are produced:
• Airspace opacity (Fig. 11.48), which may be localized or diffuse. In some instances the pattern is that of Löffler syndrome. Illnesses vary from a mild, simple eosinophilic pneumonia to a fulminant acute eosinophilic pneumonia. 266 Drugs that are particularly associated with a consolidative pattern include penicillin, sulfonamides, para-aminosalicylic acid, chlorpropamide, nitrofurantoin, methotrexate, carbamazepine, mephenesin, imipramine, trimipramine, and hydrochlorothiazide.
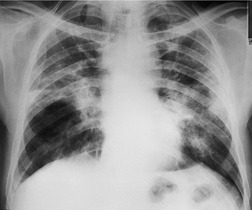 |
| Fig. 11.48 (Courtesy of Dr. J D Stevenson, Poole, Dorset, UK.) |
• Hilar lymphadenopathy. This is recorded with the antiepileptic drugs phenytoin and trimethadione. 342
• Pleural effusions. These are occasionally seen with nitrofurantoin (see p 1025).
• Reticulonodular opacity. An interstitial pulmonary fibrosis type of pattern is produced in particular by nitrofurantoin and methotrexate. With nitrofurantoin the chronic interstitial pattern is associated with a blood eosinophilia in about 40% of patients. 343 Other drugs that produce an interstitial pattern include gold342 and clofibrate. 344
• Other patterns. Patterns other than those described and which are not easy to classify are recorded with a number of drugs such as penicillamine. 345
A distinctive syndrome was described in association with l-tryptophan ingestion (l-tryptophan eosinophilia myalgia syndrome). Principal features include peripheral eosinophilia, myopathy, peripheral neuropathy, eosinophilic fasciitis, and respiratory disorder. This latter is characterized on imaging by bilateral, often basally predominant, opacity with mixed alveolar and interstitial features, and pleural effusions.346. and 347. Pathologically there is chronic interstitial pneumonitis, tissue eosinophilia, and vasculitis. 346
Tropical pulmonary eosinophilia
This is a specific systemic disease caused by hypersensitivity to microfilariae – the early larval forms of various filarial nematodes, the most important being Brugia malayi and Wuchereria bancrofti.348. and 349. Transmission is by mosquito. Tropical pulmonary eosinophilia (TPE) is found in all parts of the world where filariasis is endemic, particularly the Indian subcontinent, South East Asia, the South Pacific, North Africa, and South America. It occurs chiefly in the indigenous residents (particularly in the Indian subcontinent),350. and 351. and is very uncommon in visitors unless they have resided in the area for many months. 352 In nonendemic areas the disease is seen in immigrants and, because of persistence of the parasite in the host, may present as long as 3 years after returning from an endemic area. 353 Occurrence is extremely rare in Caucasians. 354
The disease is more common in males, in some series by as much as 4 : l, 355 but this is not a universal finding. 354 The usual age at presentation is between 5 and 40 years, 355 though the range can be larger; in one series of 350 it varied from l.5 to 74 years. 351 The principal features of the illness are a systemic disturbance marked by fatigue, weight loss, and low-grade fever together with chronic cough, which is particularly troublesome at night. Even without treatment, symptoms tend to remit after several weeks or months only to recur later. 352 Patients who have not had treatment or who have responded inadequately may go on to develop progressive dyspnea secondary to interstitial fibrosis. 349 There is a gross eosinophilia of more than 3 × 109/L, characteristically between 5 × 109/L and 60 × 109/L, IgE levels are greatly elevated, usually more than 1000 units/mL, and there is a high titer of antifilarial antibody. BAL shows a high percentage of eosinophils, in the order of 50%.
Imaging findings in the chest351.354.356. and 357. may be caused by active alveolitis and/or interstitial scarring. The chest radiograph has a normal appearance in 2–13% of cases. The most common abnormalities, seen in 30–60% of patients, are fine linear opacities distributed diffusely and symmetrically, accompanied by hilar haziness, and an accentuation or blurring of vessels. Some authors354 describe a basal preponderance, and others stress a diffuse ground-glass type of pattern. 351 Small nodules are a slightly less common finding, seen in about 30–50% of patients. The nodules range in size from l mm to 5 mm and may occur alone or with the linear opacities described above. 354 Though generally bilateral and symmetric, nodules may be asymmetric or even unilateral. Other patterns are much less common and consist of areas of consolidation that are generally small and single. In a study of 10 patients with tropical pulmonary eosinophilia, 358 CT showed reticulonodular abnormality, bronchiectasis, and air-trapping, but the extent of CT abnormalities did not correlate with physiologic impairment.
Tropical pulmonary eosinophilia characteristically responds rapidly to diethylcarbamazine therapy and this is an important diagnostic criterion. However, 5% of patients, even early on in the natural history of the disease, fail to respond adequately and this figure rises to 20–40% in patients who have had longstanding symptoms. 349 A 5-year follow-up of a large series of patients showed a 20% relapse rate349 and a significant number of patients develop chronic, progressive dyspnea on the basis of interstitial fibrosis.
Eosinophilic lung disease from other worm infestations
The larval stages of a number of worms other than filarial nematodes pass through the lung and may, in the process, induce an allergic response. This most commonly takes the form of simple eosinophilic pneumonia/Löffler syndrome with transient, migratory, nonsegmental areas of consolidation associated with a blood eosinophilia (Fig. 11.49). 359 Nearly all the worms that cause this response are nematodes: Ascaris lumbricoides,360Ascaris suum,361Strongyloides stercoralis, 359Toxocara canis (visceral larva migrans), 362Toxocara cati, 359Ancylostoma braziliense (‘creeping eruption’), 363Ancylostoma duodenale, Necator americanus, 364 and Trichuris trichiura. 242 Exceptions are Taenia saginata and Echinococcus alveolaris, 242 both cestodes, and Schistosoma spp., which are trematodes. 365 It is possible that, in at least some of these infestations, the pulmonary reaction is not related to local larvae but rather is a remote response to a soluble antigen. 363 Thus in 26 patients with eosinophilic lung disease due to Ankylostoma braziliense (Fig. 11.49), it was not possible to demonstrate larvae in the sputum. 366 Some idea of the possible frequency of eosinophilic lung disease caused by nematode infestations may be gained from the last authors, who found lung parenchymal abnormality in 34% of 76 patients with cutaneous larva migrans (hookworm infestation).
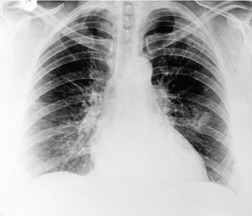
In the USA, the commonest parasites to consider are Strongyloides, Ascaris, Toxocara, and Ancylostoma. 266 The last three agents usually give rise to simple eosinophilic pneumonia/Löffler-type changes on the chest radiograph. With Strongyloides the lung pathology is more complicated and in the autoinfection stage there is a pulmonary foreign body reaction with inflammatory pneumonitis, bronchopneumonia, and hemorrhage. The chest radiograph (Fig. 11.50) reflects this and shows mixed opacity – miliary nodules, reticular opacities, and airspace opacities, ranging from multifocal and patchy to lobar. 367 About 90% of patients will have a blood eosinophilia. Should the hyperinfection syndrome supervene, the chest radiograph manifests widespread consolidation, at which stage peripheral eosinophilia may be absent. 367
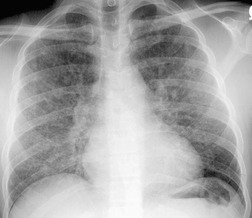
It is important to know that, for a variety of reasons, acute eosinophilic lung disease induced by Strongyloides, Ascaris, Toxacara, and Ancylostoma is usually not accompanied by ova in the stool.241. and 266. Ascariasis and Strongyloides may be diagnosed by finding larvae in the sputum, BAL, or gastric aspirates, but ova may not be found in the stool until females are mature, which in ascariasis may be delayed up to 3 months after initial pulmonary manifestations. 241Toxocara and Ancylostoma do not replicate in the gut, so ova are not usually found in the stool. Both conditions may be suspected on the basis of a skin rash; toxocariasis may be confirmed by serology. 241
Bronchocentric granulomatosis
This uncommon form of pulmonary granulomatosis, first described by Liebow in 1973, 368 differs from other lung granulomatoses, such as Wegener granulomatosis, in that it is localized to the lung, and centered around airways (bronchocentric) rather than vessels (angiocentric). Pathologically, small airways and bronchioles are filled and replaced by cellular debris and necrotic granulomas surrounded by palisaded epithelioid cells. At least 30% of cases are related to asthma, and in these individuals is probably part of the spectrum of ABPA.369. and 370. In asthmatic people, the major part of the cellular infiltrate is made up of eosinophils, whereas in nonasthmatic people the plasma cell is dominant. 371 Large airways may show bronchocele formation, and distal lung is often consolidated by an eosinophilic or obstructive pneumonitis. Vasculitic changes appear to be minor and incidental.
Only four series of patients with bronchocentric granulomatosis have been recorded,371.372.373. and 374. representing a total of 60 cases. However, there have been many additional case reports. Patients commonly present in their forties, but there is a wide age range (9–76 years) and a tendency for asthmatic patients to present at a younger age (mean age 22 years) than nonasthmatic patients (mean age 50 years). 371 The incidence in both sexes is equal. Symptoms may be absent or minor and, when present, consist of fever, cough, chest pain, wheeze, and hemoptysis. About 50% of patients have had a blood eosinophilia, a finding that appears to be limited to asthmatic people.371. and 373.
On imaging,371.372.373. and 375. two major patterns are seen: consolidation and masslike lesions. Consolidation may be lobar or sublobar and may be accompanied by volume loss (Fig. 11.51). They are thought to represent either eosinophilic or obstructive pneumonitis. 371 They tend to be more common in the upper zones,371. and 375. and are unilateral in about 75% of patients. Sublobar consolidation was the commonest finding (16 out of 22) in one large series. 371 Masslike lesions (Fig. 11.52) are commonly solitary, but can be multiple. They are considered to represent a mass of necrotic tissue with surrounding granulomatous or organizing pneumonia. They vary in size from 2 cm to 15 cm, 376 and are often not particularly well defined. They may be centered on an airway. Occasionally they cavitate.371.376. and 377. Less common imaging patterns include mucoid impaction368.371.377. and 378. and reticulonodular opacities. 377 On some occasions the reticulonodular abnormality has evolved from antecedent consolidation. Adenopathy and pleural disease are not features. 377 In a study of five patients with bronchocentric granulomatosis, 374 CT showed spiculated mass lesions in three cases and consolidation in two. Extensive mucoid impaction was seen in two patients. The imaging findings were felt to be due to granuloma formation with or without proximal airway obstruction.
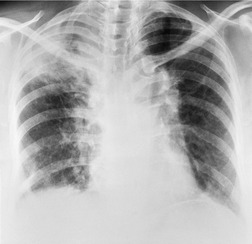
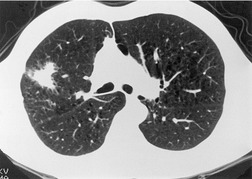
Prognosis is good. Lesions may clear spontaneously or with steroids and generally do not recur following surgical removal. If this should happen, recurrences can usually be controlled with steroid treatment.
Hypereosinophilic syndrome
Hypereosinophilic syndrome is an uncommon heterogeneous group of disorders in which the eosinophilia may be neoplastic (such as an eosinophilic leukemia) or reactive, and in the latter instance some appear to be truly idiopathic in that a cause is not established even at death. 379 It is characterized by: (1) prolonged and marked eosinophilia (a blood count of more than 1.5 × 109/L for more than 6 months); (2) no recognizable cause for the eosinophilia such as parasitic infestation or allergy; and (3) signs or symptoms of organ dysfunction (Box 11.11). 380 The sustained hypereosinophilia causes tissue damage, particularly cardiac, and this is a key element to the syndrome. The illness varies in severity from mild to fatal, involving in particular the cardiovascular and nervous systems. The cardiac abnormality consists primarily of endocardial thickening and fibrosis related to widespread tissue infiltration with mature eosinophils, 381 leading to endocardial fibrosis, restrictive cardiomyopathy, and thrombosis. Several subtypes of hypereosinophilic syndrome have been described, including myeloproliferative, lymphocytic, and familial variants. 382 A significant recent advance in understanding of this condition has been the recognition of FIP1L1-platelet-derived growth factor receptor-α fusion protein in up to 50% of individuals with this syndrome, and the recognition that these individuals may respond to treatment with the protein kinase inhibitor imatinib.245. and 382. Other forms of this condition may respond to treatment with mepolizumab. 383
Box 11.11
• Cardiac
– Endocardial thickening
– Restrictive cardiomyopathy
• Skin rash
– Pulmonary parenchymal opacity
Almost all patients have been men, typically young or middle-aged adults who present with progressive cardiopulmonary symptoms, skin rash, or myalgia, together with systemic symptoms such as weight loss, weakness, fatigue, and fever. The peripheral blood shows a marked eosinophilia, often in the order of 30–70% of the total white blood cell count (10–50 × 109/L), some of the eosinophils being degranulated and vacuolated. Some patients have a mild increase in IgE. The organ systems most commonly affected are the nervous and cardiovascular systems and the skin. Cardiovascular involvement is usually the dominant feature and is a major cause of morbidity, with signs of restrictive cardiomyopathy and pump failure, mitral and tricuspid regurgitation, and endocardial thrombosis. The last leads to systemic emboli in about 5% of patients384 or pulmonary emboli if the thrombus is right sided. 381
Clinical pulmonary involvement has been recorded in about 40% of patients.380. and 381. In most, these are findings related to heart failure, 385 and include pulmonary edema (Fig. 11.53) and pleural effusions. 380 Because the cardiomyopathy is restrictive in type, any accompanying cardiomegaly is often mild. 384 Pulmonary emboli were recorded in 9% of 57 patients reviewed from the literature. 380 Other less common pulmonary manifestations include transient consolidations, which are presumably eosinophilic pneumonias and diffuse interstitial fibrosis.380. and 384. HRCT findings in five patients in whom chest radiographs were normal (3/5) or showed nonspecific focal opacities (2/5) consisted of scattered nodules (about 2–10 mm) with ground-glass halos (5/5) and focal areas of ground-glass attenuation (3/5) (Fig. 11.53). There was no pathologic correlation, but the authors considered that the lesions probably represented areas of eosinophilic infiltration. 386 In three patients studied by Johkoh et al., 262 the common imaging features were ground-glass attenuation and nodules, with septal thickening and thickening of bronchovascular bundles. The challenge in these cases is to distinguish findings due to heart failure from those due to pulmonary involvement.
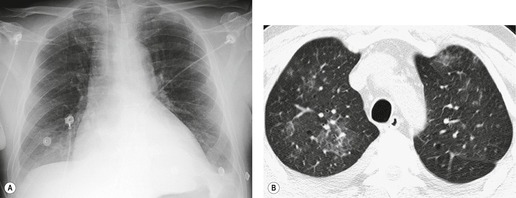
Other conditions associated with eosinophilia (Box 11.12)
There is often a mild to moderate eosinophilia in patients with asthma. 388 The radiology of asthma is discussed elsewhere (p 752). In addition, asthma is often a prominent symptom in a number of specific eosinophilic lung states such as ABPA and the Churg–Strauss syndrome. Other pulmonary conditions associated with blood eosinophilia, and often with tissue eosinophilia, are listed in Box 11.12. The degree of eosinophilia in these conditions is generally relatively slight (less than 1 × 109/L). Box 11.12 emphasizes the importance of considering infection and neoplasm (Fig. 11.54) in patients presenting with eosinophilia and pulmonary parenchymal abnormalities.
Box 11.12
• Asthma
• Hyperimmunoglobulin E syndrome
• Langerhans cell histiocytosis
• Infections
– Bacterial (Brucella, Mycobacterium)
– Chlamydia
– Viral (adenovirus)
– Protozoal (Pneumocystis, Triomonas)
– Fungal (Coccidioides, Histoplasma)
• Neoplasms
– Bronchogenic carcinoma, bronchial carcinoid
– Metastases
– Irradiated neoplasms
– Lymphoma (Hodgkin and non-Hodgkin, lymphomatoid granulomatosis)
• ‘Immunologic’ conditions
– Wegener granulomatosis
– Rheumatoid disease
– Hypersensitivity pneumonitis
– Sarcoidosis
– Idiopathic pulmonary fibrosis
– Cryptogenic organizing pneumonia
• Miscellaneous
– Hemodialysis
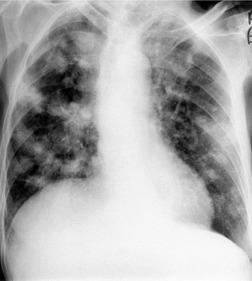
Hyperimmunoglobulin E syndrome
This rare primary immunodeficiency syndrome was first described in 1966389 and is characterized by recurrent bacterial infections of the lungs, sinuses, and skin dating from birth or early childhood, and a more than 10-fold elevation of serum IgE. 390 The immunologic derangement is complex and only partly understood. 391 The number of suppressor T cells is reduced, and this appears to result in increased production of IgE antibodies to staphylococcal antigens. A neutrophil chemotactic defect may also be present. 392 Recurrent bronchitis and pneumonia is a major feature, often due to Staphylococcus aureus though other bacteria and fungi may also be responsible. There is commonly an eczematous dermatitis, mucocutaneous candidiasis, recurrent furunculosis, and cutaneous cold abscesses. The lack of the usual systemic and local inflammatory findings with the abscesses is a striking feature, particularly because most are due to Staphylococcus aureus. Pneumonia and cystic lung disease is the most common cause of death, and Aspergillus and/or Pseudomonas infection is usually found at autopsy. 393 Mild or mild to moderate eosinophilia occurs in 77–100% of patients.390. and 391. Occasionally, however, eosinophilia is marked. 394
Imaging findings in the chest391.392. and 395. consist of recurrent infective consolidation (beginning before the age of 3 years) and cyst formation. All the patients in the series of Merten and co-workers, 391 in which the average age was 18 years, had lung cysts. Some cysts disappeared after a few years while others were recurrent or persistent. About a third of cysts were multiple and could be very large, occupying much of a hemithorax, while their walls were usually smooth but of varying thickness. The cysts probably represent the residue of lung abscesses or pneumatoceles. 393 Other pulmonary findings include empyema, fungal superinfection of pneumatocele, 396 bronchopleural fistula,392. and 397. bronchiectasis,391. and 396. and pneumothorax.392.396. and 398. Eighty percent in Merten’s series had chronic sinusitis. 391
PULMONARY ALVEOLAR PROTEINOSIS
Pulmonary alveolar proteinosis (PAP) is a rare disorder first described in 1958399 and characterized pathologically by alveolar filling with a lipid-rich, proteinaceous material (positive to periodic acid–Schiff stain), while the lung interstitium remains relatively normal. Its incidence is 0.2–0.5 new cases per 1 000 000 persons each year, and its prevalence is 2–6 cases per 1 000 000 persons.400. and 401. PAP may be divided into idiopathic and secondary forms (Box 11.13). 401 Idiopathic PAP accounts for about 90% of all cases.
Box 11.13
Idiopathic
Secondary
• Dust/chemical exposure
– Silica
– Aluminum
– Titanium
• Infection-related
– Pneumocystis
• Hematologic/immunocompromise
– Lymphoma
– Myeloid leukemia
– Severe combined immune deficiency
– Lung transplant
• Medications
• Congenital
– Surfactant apoprotein deficiency
• Familial
Secondary forms are seen in association with:
• Infections. The association is complicated and some infections (see below) are undoubtedly secondary to primary PAP whereas other infections are themselves probably the primary process that precipitates the proteinosis. An example of the latter is pneumocystis infection, both HIV and non-HIV related. 409
• Hematologic malignancy or immunocompromise. An association with hematologic malignancies (lymphoma, myeloid leukemia, myelodysplastic syndrome) is described, particularly in children.410.411.412. and 413. In another review 30% of children with PAP had thymic alymphoplasia. 414 PAP is also described with severe combined immune deficiency (SCID) in both mice415 and humans. 416 Three cases of PAP developing in lung allograft recipients performed for conditions other than PAP have been recorded. 417
The intraalveolar lipoproteinaceous material in PAP consists primarily of surfactant and associated apoproteins. 421 Both congenital and primary PAP are associated with deficiency or impaired function of granulocyte–macrophage colony-stimulating factor (GM-CSF), which regulates surfactant homeostasis and immune defense. 401 Patients with idiopathic PAP all have high levels of autoantibodies against GM-CSF, which neutralize the role of GM-CSF in the differentiation of alveolar macrophages, thus critically impairing the process of surfactant clearance in the lung. 401 These antibodies also appear to neutralize the antimicrobial activity of GM-CSF by inducing lung neutrophil dysfunction.425. and 426. Knockout mice with deficiencies of the GM-CSF, or the β-GM-CSF receptor all develop PAP. 427
PAP is most common in adults, particularly between 30 and 60 years of age, although cases are seen in children (including neonates) and up to the age of 72 years.400. and 427. Sixty to eighty percent of patients are male.400.402.426. and 427. About 50–70% are cigarette smokers, and the male predominance may be due to the higher frequency of cigarette smoking in males.400. and 427. In children, PAP is usually associated with surfactant deficiency or immunocompromise, and the disease is progressive and often fatal, whereas adults generally have no underlying disorder. In about one-fifth of cases the onset is acute, with fever, weight loss, and dyspnea, either with or without a superadded opportunistic infection. 402 Most of the other cases have an insidious onset with progressive dyspnea and cough. Median duration of symptoms prior to presentation is 7 months. 427 Cough is usually dry but is sometimes productive of white sticky sputum. Other features include pleuritic chest pain, hemoptysis, 426 and pneumothorax. 402 The clinical signs consist of crackles and occasionally clubbing. PAP is one of the conditions in which symptoms and clinical signs may be mild in the face of striking imaging signs (‘clinicoimaging discrepancy’).401. and 428. In up to 30% of cases the disease is discovered by chest radiography in asymptomatic or minimally symptomatic individuals. 400
Seventy percent of patients have a mild to moderate elevation of serum lactate dehydrogenase (LDH). The serum levels of surfactant apoproteins A and D (SP-A, SP-D) are markedly elevated, though nonspecific. 429 Antibodies to GM-CSF may be diagnostically useful. Respiratory function tests show a restrictive defect with hypoxemia and impaired diffusion. 426 Diagnosis is established by BAL showing the characteristic eosinophilic, granular, PAS positive lipoproteinaceous material in the washings. 429 A recent report suggests that elevated levels of the apoprotein SP-D in the washings may be specific for PAP. 430 In problematic patients it may occasionally be necessary to obtain a transbronchial lung biopsy for histologic study. 429
On the chest radiograph, the main findings are consolidation ground-glass abnormality, due to alveolar filling. 399 The classic imaging finding is bilateral, symmetric consolidation, or, more commonly, ground-glass abnormality, particularly in a perihilar or hilar and basal distribution (Fig. 11.55).399.402. and 431. This opacity often has a fine granularity and air bronchograms are unusual. At other times the pattern consists of rather coarse (5 mm), ill-defined acinar nodules, perhaps in part confluent. 432 In some cases, septal thickening or reticular abnormality may be seen (Fig. 11.56).399.402.433. and 434. Although usually symmetric, the consolidation can be asymmetric, 433 unilateral,399. and 426. or lobar.399. and 410. Sometimes the consolidation is basally predominant, 410 peripheral rather than central, 428 and multifocal rather than diffuse. 426 Lung apices and costophrenic sulci are usually spared, and a thin zone of clear lung immediately above the diaphragm is characteristic, perhaps because diaphragmatic movement helps squeeze proteinaceous material out of the adjacent airspaces. Simultaneous evolution and regression, producing a shifting pattern similar to that found in Löffler syndrome, has been described. 435
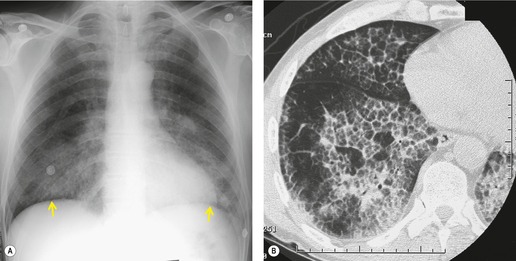
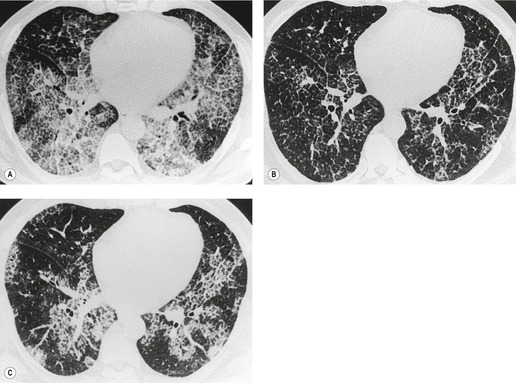
• Geographic ground-glass abnormality
• Lines perpendicular to the pleura and forming polygonal structures, representing thickened interlobular septa
• A finer network of intralobular lines
• Absence of architectural distortion or traction bronchiectasis.
The superimposition of the reticular pattern formed by thickened interlobular septa and intralobular lines on a background of ground-glass abnormality produces the characteristic ‘crazy-paving’ pattern (Figs 11.55 and 11.56). Imaging changes tend to be diffuse,437. and 440. but may predominate in any lung region or zone.401. and 440. Involved areas are bordered by normal lung and have a sharp, geographic margin reflecting lobular boundaries. 401 In an AFIP series, 25% of cases were characterized by ground-glass abnormality without crazy-paving pattern. 401 Other airspace opacities that may be found include acinar nodules, limited air bronchograms, and areas of consolidation where the density of the opacity obscures underlying vessels. If consolidation is present, the possibility of superimposed infective consolidation should be considered.
The ground-glass abnormality is due to accumulation of protein within the alveoli, and the interlobular and interlobular lines are due to expansion of interlobular septa by edema and markedly dilated lymphatics. 401 Unusual cases of PAP may be associated with histologic lung fibrosis, indicated by traction bronchiectasis and fissural or architectural distortion. 401
Following therapeutic whole-lung lavage the ground-glass opacity decreases440 (Fig. 11.56) and the reticulation may become more prominent. 437 The crazy-paving pattern has been described in a wide variety of lung diseases,441. and 442. including exogenous lipoid pneumonia, 443 bronchioloalveolar cell carcinoma, 444 drug toxicity, sarcoidosis, nonspecific interstitial pneumonia, organizing pneumonia, pulmonary hemorrhage, Pneumocystis jirovecii pneumonia, and ARDS. The crazy-paving pattern of PAP may distinguished from these other entities in the following ways: clinical presentation is indolent; extent of imaging abnormality is out of proportion to clinical symptoms and physiologic impairment; architectural distortion and traction bronchiectasis are typically absent; distribution is geographic; and thickened interlobular septa are more prominent than in the other conditions mentioned above. Where PAP is the leading imaging diagnosis, it is appropriate to recommend BAL to confirm the diagnosis and exclude other entities such as infection, hemorrhage, bronchioloalveolar cell carcinoma, and lipoid pneumonia.
Because of the functional macrophage and neutrophil impairment, complicating infections by pathogenic bacteria431 or opportunistic bacterial, fungal, and viral410 agents are common. In one review, 445 15% of 160 patients had such infections, and they were a major cause of death402 before therapeutic lavage was introduced. 426 Agents most commonly recorded are Nocardia,402.410. and 433. Mycobacterium tuberculosis and nontuberculous mycobacteria,446. and 447. and Cryptococcus. 446 It seems likely that nocardial infections have become less frequent following the adoption of therapeutic lavage. 429 Radiographic pointers to such an opportunistic infection include the development of focal consolidation, cavitation, and pleural effusion. 402 CT may identify focal pneumonia not apparent on chest radiographs. 436
PAP improves spontaneously in about 10% of patients.402.426. and 427. Most of the remaining patients will require treatment, and the standard treatment up to recently has been with unilateral or bilateral saline whole-lung lavage. 448 Lavage has virtually eliminated mortality449 and results in complete remission in 75% of patients. 426 Chest radiograph or chest CT may be used to decide which side should be treated and to detect any complications. 450 Recurrence of PAP has been documented in a patient who had a double lung transplant for progressive disease. 451 Recently, small-scale clinical trials of GM-CSF have resulted in clinical improvement in 35–70% of cases, but the value of this treatment remains unclear.427. and 452.
PULMONARY ALVEOLAR MICROLITHIASIS
Pulmonary alveolar microlithiasis was first described in 1918. 453 It is a rare disorder, with 225 cases recorded in the world literature. 454 A recent advance has been the identification of a mutation in the SLC34A2 gene, which encodes a phosphate transporter gene, in all of 13 patients in two studies.455. and 456. This gene may also be associated with testicular microlithiasis. 455
Alveolar microlithiasis is characterized pathologically by the accumulation of numerous, largely intraalveolar, calcified bodies (calcispherites or microliths). Microliths have a mean diameter of about 200 µm457 and contain calcium phosphate. 369 Dystrophic ossification occasionally develops around microliths. 457 There have been rare descriptions of extrapulmonary microliths in the testes or kidneys.458.459. and 460. Alveolar walls are commonly normal461 but later in the disease interstitial fibrosis may develop together with bullae and blebs.460. and 462.
Although alveolar microlithiasis occurs worldwide, nearly a quarter of recorded patients are from Turkey. 454 About 40% of cases are sporadic, while the remainder show a strong familial association, 462 consistent with an autosomal recessive transmission.463. and 464. Other congenital disorders are occasionally associated such as Waardenburg anophthalmia syndrome464 and diaphyseal aclasia. 465 There is no gender difference in incidence. 461 The disease is commonly first detected in the third and fourth decades, 461 but the range is large, with the disorder recorded in premature neonates466 and in an 80-year-old woman. 460
Like PAP, gross radiographic changes are often present in the face of minor clinical symptoms. In an analysis of published papers comprising 576 subjects, 50% were asymptomatic at the time of diagnosis. 467 The interval between radiographic diagnosis and onset of symptoms may be from 5 to 40 years. 463 Later in the course of the disease cough may appear, and in a small proportion of patients hemoptysis, clubbing, hypertrophic osteoarthropathy, 454 pneumothorax, dyspnea, and cor pulmonale develop.461. and 462. Respiratory function tests have been abnormal in about a third of reported cases and most commonly show a restrictive defect or a decreased carbon monoxide diffusing capacity. 461 Identification of microliths in expectorated sputum or BAL can be diagnostic,468. and 469. and whole-lung lavage has been used to relieve symptoms of dyspnea. 468
The chest radiograph is characteristic, 462 with innumerable, widespread, pinpoint nodules of calcific density. Nodules are less than 1 mm in diameter, but they may summate in areas to give a ground-glass, reticular, or more coarsely nodular (up to 5 mm) pattern (Fig. 11.57). 470 The radiographic opacity, which tends to be greatest at the bases, sometimes shows subpleural462.470.471. and 472. or peribronchovascular accentuation. 470 In advanced disease the radiographic opacity is so great that anatomic landmarks become completely obscured. Thus the heart may ‘vanish’470 or even appear as a transradiant area on a penetrated radiograph. The parietal pleural region may appear as a thin dark band (Fig. 11.57), 462 which is shown by CT to be due to subpleural cysts or paraseptal emphysema.473.474.475. and 476. Pleural fissures can appear dense, thickened (Fig. 11.57), and beaded, 477 and septal lines are occasionally seen.470. and 478. Bullae and blebs develop with late fibrosis,460.461. and 462. and these are particularly well seen on CT.457. and 479. Such blebs and bullae probably predispose patients to pneumothorax and pneumomediastinum, which are recognized complications.460. and 479.
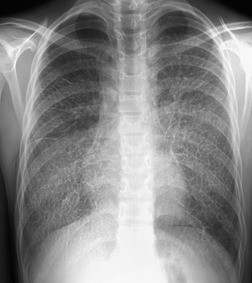
On CT, the predominant distribution of abnormality is usually posterobasal (Fig. 11.58).457.464.471.473.474.475.476.479.480.481. and 482. Because of volume averaging of the tiny microliths, their calcific density may not be apparent, resulting in a micronodular or ground-glass pattern, even on thin-section CT. With larger nodules, the calcification may be visible. The occasional 5 mm nodule presumably represents dystrophic ossification seen histopathologically. Ground-glass abnormality is the most common pattern, seen in nine out of 10 patients in a Brazilian study. 476 Calcification may be uniform, producing dense consolidation (seen in 50%) (see Fig. 3.118B), or may be linear or nodular. Linear calcification may be subpleural, with nodular fissural thickening, or along interlobular septa or bronchovascular bundles (Fig. 11.58).464.471.473.474.479. and 482. Crazy-paving pattern may be seen. 483 All CT studies showed airspaces (blebs or bullae) either apically,473.474.479. and 480. or throughout the lung, or subpleurally.471.473. and 480. The cysts represent dilated alveolar ducts. 484 A striking subpleural peel of paraseptal emphysema may be seen.473. and 475.
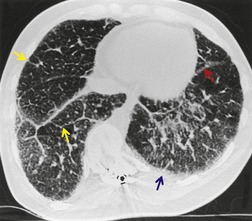
There is a single case report of MR findings in alveolar microlithiasis. 480




Related posts:
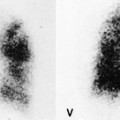
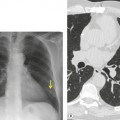
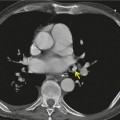
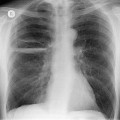
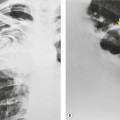
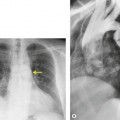
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
