Inhaled agents may injure any part of the tracheobronchial tree or the lung parenchyma: the site of injury depends on the physical characteristics of the agent (especially its physical state and particle size). Many inhaled agents will affect more than one compartment of the lung. For instance, inhaled cigarette smoke, the commonest cause of inhalational lung injury, may cause tracheitis, bronchitis, bronchiolitis, and parenchymal inflammation and destruction. Other inhaled agents include mineral dusts that cause pneumoconiosis, organic agents causing hypersensitivity pneumonitis, noxious fumes, and aspirated fluids or solids.
SMOKING-RELATED LUNG DISEASES
Respiratory bronchiolitis/respiratory bronchiolitis interstitial lung disease
Respiratory bronchiolitis (RB) is a histopathologic lesion found in cigarette smokers, characterized by the presence of pigmented intraluminal macrophages within first- and second-order respiratory bronchioles. It is usually asymptomatic, and the histologic finding of RB is present at autopsy in virtually all cigarette smokers dying of incidental causes. 1 The prevalence of this histologic finding decreases after smoking cessation: in one study, it was found in a third of patients 5 years after discontinuing smoking, and in one patient who had stopped 32 years before. 2 The histologic pattern of RB is virtually specific for cigarette smoking: although inflammation and fibrosis of the respiratory bronchioles may occur in individuals exposed to environmental particulates, the histologic pattern of this form of RB is quite different from that found in smoking-related RB. 3
There is increasing evidence that smoking-related lung disease may be visible on CT in nonsmokers with exposure to second-hand smoke. Typical histologic and imaging features of RB, with normal pulmonary function, have been described in a nonsmoker with occupational exposure to second-hand smoke. 4 In a multivariate analysis of 310 asbestos workers who were never smokers or former smokers, the extent of ground-glass abnormality and irregular linear opacities was significantly related to the intensity of domestic or occupational exposure to second-hand smoke. 5
In contrast to asymptomatic RB, respiratory bronchiolitis interstitial lung disease (RB-ILD) is a clinical diagnosis made in heavy smokers when respiratory bronchiolitis is extensive enough to cause symptoms and physiologic evidence of interstitial lung disease.6.7.8.9. and 10. RB-ILD usually affects heavy smokers, with average exposures of over 30 pack years of cigarette smoking. Although the CT appearances of RB, RB-ILD, and desquamative interstitial pneumonia (DIP) overlap (Table 8.1),11. and 12. and the pathologic findings may coexist, DIP should probably be regarded as a distinct entity, because of its different histologic appearance, inconstant relationship to cigarette smoking, 13 and less benign prognosis. 10 DIP may also be seen in individuals with surfactant deficiencies.14. and 15.
| Symptoms | Physiologic impairment | Pathologic findings | Imaging features | |
|---|---|---|---|---|
| RB | Minimal or absent | None | Intraluminal pigmented macrophages | Centrilobular nodules, small areas of ground-glass attenuation |
| RB-ILD | Marked | Marked | More extensive peribronchiolar macrophages and interstitial thickening | Widespread centrilobular nodules and ground-glass appearance |
| DIP | Marked | Marked | Diffuse intra-alveolar macrophages and interstitial thickening | Widespread ground-glass abnormality, ± cysts |
| Pulmonary Langerhans cell histiocytosis | Variable | Marked | Peribronchiolar accumulation of Langerhans cells | Poorly defined nodules, cavities, irregular cysts |
On the chest radiograph, the findings of smoking-related RB or RB-ILD are subtle, nonspecific, and difficult to describe or illustrate. The pattern has been variously described as reticular, reticulonodular, or ground-glass pattern.6.9.16. and 17. Some have used the term ‘dirty lung’18 to describe the prominence of bronchovascular structures, and slight blurring of bronchovascular outlines commonly seen in heavy smokers. This parenchymal abnormality is probably due to a combination of thickening of the walls of bronchi and bronchioles, emphysema, and patchy ground-glass abnormality of RB-ILD. 18
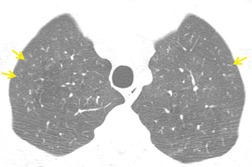
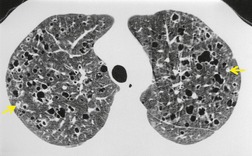
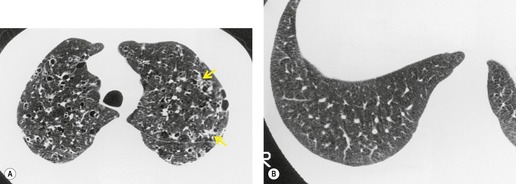
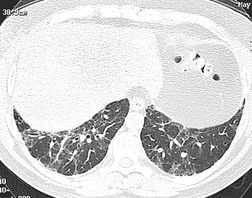
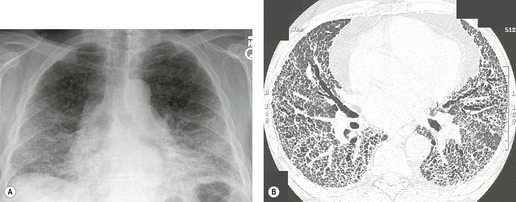
On computed tomography (CT), patients with asymptomatic RB generally show mild centrilobular nodularity and small patches of ground-glass attenuation (Fig. 8.1). 19 Remy-Jardin et al., 20 in a longitudinal study of smoking-related CT changes, found that in patients with centrilobular micronodules who continued to smoke, the centrilobular nodules most commonly either increased in profusion or evolved into areas of emphysema. Emphysema and ground-glass abnormality also increased in prevalence in those who continued to smoke. The presence of emphysema and/or ground-glass abnormality on a baseline scan was associated with a significantly more rapid decline in physiologic indices of airway obstruction.
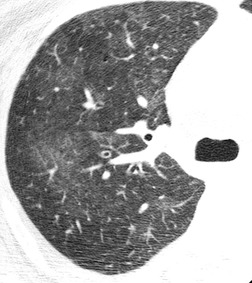
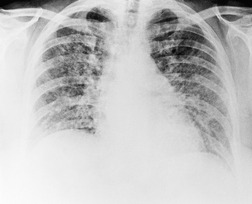
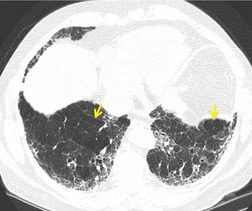
In RB-ILD, centrilobular nodularity and ground-glass attenuation are usually more extensive than in asymptomatic RB (Fig. 8.2). 16 Emphysema, bronchial wall thickening, and areas of decreased lung attenuation are commonly also present. 17 When patchy abnormalities of this type are present in heavy smokers with impaired pulmonary function, RB-ILD may be diagnosed with a high degree of confidence. The CT findings of RB-ILD are at least partially reversible in patients who stop smoking.17. and 21.
The CT features of RB-ILD may be similar to those of hypersensitivity pneumonitis, DIP, and nonspecific interstitial pneumonia (NSIP). RB-ILD differs from DIP in that the ground-glass attenuation of RB-ILD is usually less extensive, more patchy, and more poorly defined than in DIP. Centrilobular nodules are uncommon in DIP. However, RB-ILD with extensive ground-glass abnormality may be indistinguishable from DIP and HP. Clinical differentiation of RB-ILD from HP is facilitated by the fact that most patients with HP are nonsmokers.22. and 23. Sharply defined lobular air-trapping on CT is suggestive of HP rather than DIP or RB-ILD.
Desquamative interstitial pneumonia
DIP was one of the first conditions to be differentiated histologically and prognostically from usual interstitial pneumonia (UIP).24.25. and 26. Histologically, it is characterized by diffuse accumulation of large cells within the alveoli, previously thought to be desquamated pneumocytes, but now known to represent alveolar macrophages. 27 There is a variable amount of associated inflammatory change in the alveolar walls. Typically, the disease is relatively homogeneous at a microscopic level, with little lung fibrosis. It usually occurs in cigarette smokers, and is thought to represent an idiosyncratic response to inhalation of cigarette smoke. Unlike most inhalational lung diseases, it does not have a peribronchiolar predominance, and therefore is characterized on CT by a diffuse ground-glass pattern, rather than centrilobular nodules.
The clinical presentation of DIP is similar to that of the other interstitial pneumonias, such as UIP and NSIP. 25 Exertional breathlessness and a nonproductive cough are the most common symptoms.
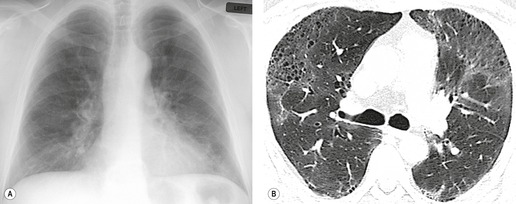
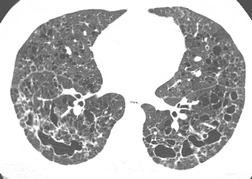
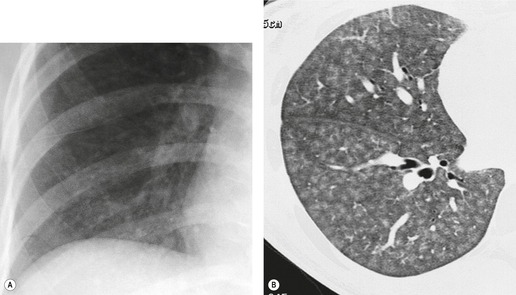
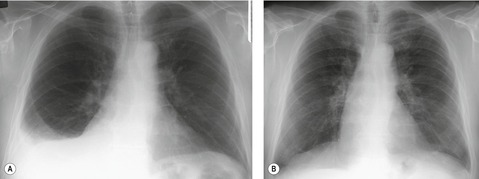
On the chest radiograph, the findings of DIP are relatively nonspecific, with reticulonodular abnormality or ground-glass attenuation being the salient features28 (Fig. 8.3). The chest radiograph may be normal in up to 20% of cases. 29 On CT, ground-glass attenuation is seen in all patients with DIP, 30 with lower lobe predominance in about 75% of the patients (Fig. 8.4). Peripheral predominance is seen in about 60%. Signs of lung fibrosis with anatomic distortion are seen in about 50% of cases, but this is not usually a dominant feature. Micronodules are seen in a small number of patients, perhaps a reflection of associated respiratory bronchiolitis. An unusual feature seen in some patients with DIP is the presence of small cysts (Fig. 8.3), which differ from honeycomb cysts in that they are not clustered and do not occur in areas of underlying fibrosis. 31 Some of these cysts resolve with time. The pathologic basis of these cysts is unclear, but they may represent dilated alveolar ducts or alveoli. 10
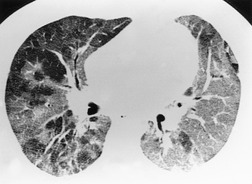
When patients with DIP are followed over time, the ground-glass attenuation usually regresses or stays stable in response to treatment. Progression to a UIP pattern is uncommon and any background, or associated interstitial fibrosis, tends to be the NSIP subtype.26. and 31. However, in contrast to the benign prognosis of RB-ILD, patients with DIP may progress to death or lung transplantation. 32
Pulmonary Langerhans cell histiocytosis
Pulmonary Langerhans cell histiocytosis (PLCH) (previously called histiocytosis X or eosinophilic granuloma of the lung) is an idiopathic disorder characterized by abnormal proliferation of the Langerhans cell, a mononuclear cell, in the lung. Histologically, the disease centers on the bronchioles, with associated involvement of the interstitium and pulmonary vasculature. 33 Histologic findings of RB and/or DIP usually coexist with those of PLCH. 34 Cellular lesions of PLCH are usually characteristic in histologic appearance, but the burnt-out phase, characterized by stellate scars, is usually more difficult to diagnose. 35
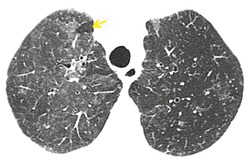
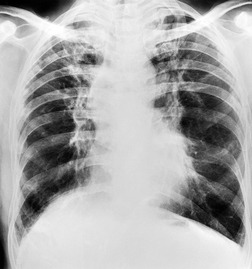
PLCH is a disease of smokers, with a history of tobacco use present in 90–100% of cases.33. and 36. Most are less than 40 years of age. Cough and dyspnea are the most common presenting complaints. 37 The presence of asymptomatic chest radiographic abnormalities occasionally precipitates the evaluation. Twenty-five percent of patients will develop a pneumothorax at some time during the course of their disease (Fig. 8.5), and this may be the presenting complaint. A few patients present with advanced pulmonary hypertension. There are several case reports and series suggesting a relationship between PLCH and malignancy, particularly hematologic malignancies and melanoma.38.39. and 40.
The physiologic abnormality of PLCH evolves over time. At presentation the pulmonary function may be normal, or may show a mixed restrictive–obstructive pattern. With progressive disease, there is an obstructive ventilatory defect with marked airflow limitation and increased lung volumes. 36 Because of the tendency of this disease to involve the pulmonary vasculature, the DLco is often markedly reduced, and gas exchange is abnormal with hypoxemia. Blood chemistry evaluation is unhelpful.
The diagnosis can sometimes be made on transbronchial biopsy or bronchoalveolar lavage, when Langerhans cells can be identified by staining for CD1a antigens. 41 A firm histologic diagnosis usually requires a surgical lung biopsy; however, surgical biopsy is now performed only in cases where the imaging appearances are atypical. The prognosis is variable, but with smoking cessation, physiologic stabilization will occur in the majority of patients.
Imaging features
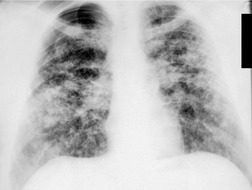
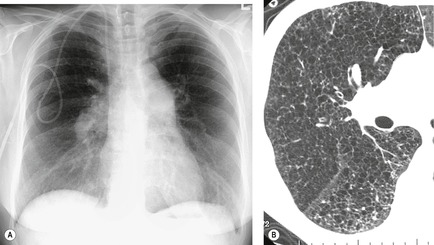
The chest radiographic appearance of PLCH is often strongly suggestive of this diagnosis (Fig. 8.6) (Box 8.1). 42 The characteristic radiographic features are a combination of cysts and nodules (the nodules are usually easier to see than the cysts). There is mid- and upper zone predominance, with sparing of the costophrenic sulci, and preservation of lung volumes. In the acute phase, the nodules are often poorly defined (Fig. 8.5), but later become well defined.
Box 8.1
• Nodules – usually poorly defined, may be cavitary
• Cysts – often irregular and odd in shape
• Upper and mid-lung predominance
• Sparing of costophrenic sulci, tips of middle lobes
• Preserved or increased lung volumes
• Pulmonary arterial enlargement at later stage
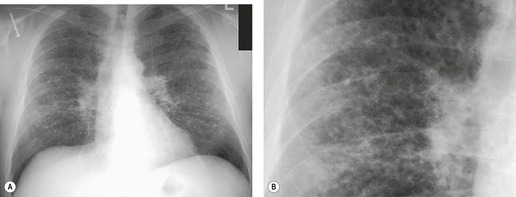
The CT appearances of PLCH are usually characteristic. Grenier et al. 43 showed that a first choice diagnosis of PLCH had a 60% likelihood of being correct when made from the chest radiograph, but had a 90% likelihood of correctness on CT scanning. The combination of pulmonary nodules and cysts is virtually diagnostic of PLCH (Fig. 8.7).42.44. and 45. Bonelli et al. 46 showed that PLCH can be confidently diagnosed in 84% of cases: confident diagnoses were correct in 100% of cases. Similar results were found in a paper by Koyama et al. 47 PLCH is usually readily distinguishable from lymphangioleiomyomatosis by the presence of nodules, by the irregular outline of the cysts and by the sparing of the lung bases. Atypical CT features of PLCH, usually requiring lung biopsy for diagnosis, may include endobronchial or tracheal lesions, solitary pulmonary nodules, and lymphadenopathy.48.49. and 50. Ground-glass abnormality may be present in some cases of PLCH, probably reflecting associated RB-ILD or DIP. 34 Associated involvement of bones, pituitary, skin, or liver may uncommonly be seen.51. and 52.
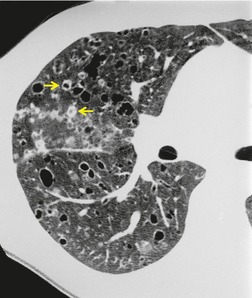
The nodules of PLCH may be poorly defined or well defined and are typically most prominent in the upper lobes (Fig. 8.7). They are usually less than 5 mm in diameter, but may be up to 1 cm. Although the nodules are usually round and poorly defined in early disease, they may be fine and irregular in later stage disease, reflecting the stellate scars identified histologically in advanced PLCH. 35 Cavities with thin or thick walls may be seen (Figs 8.7 and 8.8). 53 These are not due to necrosis, but probably result from progressive bronchiolar dilation. The cysts in this disorder are invariably well defined (Fig. 8.7) and again are usually most profuse in the upper lobes, with relative sparing of the costophrenic sulci (Fig. 8.10). 46 By three-dimensional histologic reconstruction of lesions of PLCH, Kambouchner et al. 54 demonstrated that the cavities and cysts occurring in PLCH are due to destruction of the bronchiolar wall and progressive dilation of the lumen, subsequently circumscribed by fibrous tissue, accounting for the tubular or irregular outline of these lesions (Fig. 8.7, Fig. 8.8 and Fig. 8.9). The extent of cysts, but not of nodules, in PLCH correlates with the degree of pulmonary physiologic impairment. 55 Soler et al. 56 demonstrated that the extent of nodules on CT in PLCH was correlated with active inflammatory nodules on biopsy, but also showed that even individuals with predominant cystic abnormality often had inflammatory lesions.
Regression of the chest radiographic findings of PLCH occurs in about a third of cases. 57 A study of serial high-resolution CT (HRCT) scans in 21 patients with this disease showed that the lung nodules and cavities tend to regress over time, while cysts and linear abnormalities remain the same or progress. 58 Nodules predominate on early CT scans, whereas cysts tend to predominate on follow-up scans. Parenchymal abnormality resolved completely in one of 21 patients in this study, and complete resolution has also been described by others. 59 The disease may recur in a small number of patients, usually those who continue to smoke or take up smoking again. Tazi et al. 60 described four patients who developed recurrent nodules 7 months to 7 years after initial presentation. Dauriat et al. 61 found that PLCH recurred in 20% of 39 patients who underwent lung transplantation. In a single case report, a Langerhans cell sarcoma has been reported to arise in the lungs of a patient with PLCH. 62
Patients with advanced PLCH may have marked enlargement of the pulmonary arteries and severe pulmonary hypertension (Figs 8.6 and 8.11),61. and 63. probably because Langerhans cell histiocytosis substantially involves the pulmonary vasculature.64. and 65. In one series of 21 patients with advanced PLCH, the mean pulmonary artery pressure was 59 mmHg, significantly higher than in patients with advanced chronic obstructive pulmonary disease or idiopathic pulmonary fibrosis. 66 In these patients, the CT findings may include upper lung predominant fine irregular cysts and nodules.
The CT features of pulmonary involvement in children are similar to those in adults, but mediastinal masses are more common.67. and 68. When Bernstrand et al. 69 used HRCT to evaluate patients a median of 16 years after the diagnosis of pediatric Langerhans histiocytosis, they found lung lesions (cysts or emphysema) in 10 of 41 patients, mainly in those who had multisystem involvement at presentation, in those who had received chemotherapy, and in cigarette smokers.
Other smoking-related diseases (Box 8.2)
The commonly recognized smoking-related lung diseases such as lung cancer, emphysema, and chronic obstructive lung disease are discussed elsewhere. However, cigarette smoking is also a risk factor for a number of other respiratory diseases. 70 It appears to increase the risk of respiratory infections, including the common cold, influenza, bacterial pneumonia, varicella pneumonia, and tuberculosis. 70 Smoking is associated with a greatly increased risk of pulmonary hemorrhage (Goodpasture syndrome) in patients with antiglomerular basement membrane antibodies, 71 perhaps because it causes mild alveolar injury, allowing the antibodies access to the basement membrane. Almost all patients with acute eosinophilic pneumonia are smokers (at least in the Japanese population), and this condition typically occurs within 1 month of beginning to smoke, restarting smoking, or increasing the amount of smoking.72.73. and 74.
Box 8.2
• Chronic obstructive pulmonary disease
• Lung cancer
• Idiopathic pulmonary fibrosis
• Goodpasture syndrome
• Pulmonary infection
• Acute eosinophilic pneumonia
• Spontaneous pneumothorax
Women with breast cancer who smoke have a twofold increase in the risk of pulmonary metastasis. 75 However, there is no documented increase in risk of metastases from other malignancies in smokers.
Cigarette smoking is a major risk factor for spontaneous pneumothorax, 76 and for recurrent pneumothorax. The occurrence of pneumothorax in cigarette smokers may be related to development of subpleural emphysematous blebs, but may also be related to underlying RB. 77 About 70% of patients with idiopathic pulmonary fibrosis are current or former cigarette smokers, and the odds ratio for development of idiopathic pulmonary fibrosis (IPF) in smokers is 1.6, with a higher risk for those with heavier exposures. 78 Some cases of NISP may also be smoking related.10. and 79. Smoking may also be associated with development of fibrosis in asbestos workers and patients with rheumatoid arthritis, but evaluation of this issue is confounded by the difficulty in separating the radiographic and physiologic effects of cigarette smoking from those of lung fibrosis. 70
Smokers appear to be relatively protected from development of hypersensitivity pneumonitis.23. and 80. However, smokers who develop hypersensitivity pneumonitis may have a more insidious onset, and a worse prognosis, than nonsmokers. 81 The prevalence of sarcoidosis is probably lower in cigarette smokers than in nonsmokers, 82 but its severity does not appear to differ. 83
HYPERSENSITIVITY PNEUMONITIS
Hypersensitivity pneumonitis, also known as extrinsic allergic alveolitis, is a group of pulmonary syndromes caused by repeated inhalation of and sensitization to a wide variety of organic aerosols and some chemical antigens. Diagnosis relies on a constellation of features including antigen exposure, characteristic signs and symptoms, pulmonary function abnormalities, radiologic abnormalities, and histologic findings. 84
Etiology
The extensive list of agents known to cause hypersensitivity pneumonitis can be organized into three main categories: microbial agents, animal proteins, and low-molecular-weight chemicals (Table 8.2). In a recent series of 85 consecutive cases of hypersensitivity pneumonitis presenting to the Mayo Clinic, avian antigens accounted for 29 cases (34%), with hot tub lung in 18 (21%) and farmer’s lung in 9 (11%). 85 No cause was identified in 21 cases (25%). Regardless of the antigen, typically only a minority of exposed and/or sensitized individuals will develop hypersensitivity pneumonitis.
| Agent | Source | Disease |
|---|---|---|
| Aspergillus, thermophilic actinomyces | Moldy hay, compost | Farmers’ lung, mushroom worker’s lung |
| Trichosporum asahii | Tatami mats | Japanese summer-type hypersensitivity pneumonitis |
| Mycobacterium avium complex | Hot tubs, metal working fluids | Hot tub lung, metal worker’s lung |
| Bird proteins | Bird feathers, excrement, especially doves, parakeets, feather products | Bird fancier’s lung |
| Isocyanates | Paint sprays, plastics | Isocyanate hypersensitivity pneumonitis |
As a group, various bacteria and fungi represent the most common causative agents. 84 Among bacteria, multiple species of thermophilic Actinomyces are the most frequently reported. Thermophilic Actinomyces contaminate decaying vegetable matter and are causally associated with farmer’s lung and composting (including mushroom worker’s lung).86. and 87. They may also cause disease by contaminating ventilation systems and humidifiers. 88
Nontuberculous mycobacteria contaminating hot tubs, swimming pools, or metal-working fluids are an increasingly prevalent cause of hypersensitivity pneumonitis.89.90.91. and 92. This type of hypersensitivity pneumonitis, commonly labeled ‘hot tub lung’, is probably caused by inhalation of aerosolized fluid droplets containing mycobacteria. Controversy has revolved around whether this represents infection or a true hypersensitivity pneumonitis. 89 On histology, exuberant granulomas, sometimes necrotizing, are characteristic. Although the mycobacterium usually grows from culture of lavage fluid or biopsy, the characteristic CT features of mycobacterial infection (bronchiectasis and patchy tree-in-bud pattern) are usually absent. The CT findings of ground-glass abnormality, lobular decreased attenuation, and profuse centrilobular nodules are strongly supportive of hypersensitivity pneumonitis (Fig. 8.12).93. and 94. The disease usually responds to removal of the source, with or without corticosteroid treatment.95. and 96. Antimycobacterial treatment is probably unnecessary. 96
Multiple fungi have been shown to cause hypersensitivity pneumonitis including various species of Aspergillus, Mucor, Penicillium, Basidiospores, and Trichosporon. Summer-type hypersensitivity pneumonitis, the most common form of hypersensitivity pneumonitis in Japan, is caused by Trichosporon asahii (formerly Trichosporon cutaneum). 97 Antigens from avian dander and excreta are the most common animal proteins associated with hypersensitivity pneumonitis. Causative exposures may be to live birds or to feathers in pillows or duvets.98. and 99. A review of 72 patients with bird-related hypersensitivity pneumonitis showed that the level of exposure to birds varies widely; the disease is more common in women than in men (75% vs 25%), and may occur in children. 100
Occupational exposure to isocyanate compounds is an important cause of occupational asthma and hypersensitivity pneumonitis.101.102. and 103. The chemicals are thought to combine as haptens with human proteins to form complete antigens. 87 Other rare chemical causes include acid anhydrides, pesticide pyrethrum, Pauli’s reagent (sodium diazobenzenesulfate), and copper sulfate. 84
The prevalence of hypersensitivity pneumonitis in exposed populations varies dramatically. For example, the reported prevalence of farmer’s lung has ranged from 0.02% in Scandinavia to 9% in parts of Scotland. 84 The incidence of hypersensitivity pneumonitis may be higher than this in building-related outbreaks. Environmental risk factors, including antigen concentration, duration of exposure before symptom onset, frequency and intermittency of exposure, particle size, antigen solubility and potency, use of respiratory protection, and variability in work practices may influence disease prevalence, latency, and severity. 104 Since only a small percentage of individuals with similar levels of exposure develop hypersensitivity pneumonitis, individual host factors must also play a role in the development of disease, but these remain poorly defined.
Clinical features
The clinical diagnosis of chronic hypersensitivity pneumonitis can be quite challenging because of the variety of pulmonary presentations and the diversity of potential antigenic causes, which may not be elicited in a routine medical history. 105 Classically, the presentation of hypersensitivity pneumonitis has been divided into acute, subacute, and chronic forms. However, considerable overlap between the classic presentations exists. In acute hypersensitivity pneumonitis, systemic symptoms of fever, chills, and myalgias occur along with respiratory symptoms of cough and dyspnea. These symptoms typically occur 4–12 hours after heavy exposure to the inciting antigen. The subacute and chronic forms typically present with an insidious onset of respiratory symptoms often with nonspecific systemic symptoms such as malaise, fatigue, or weight loss. A temporal relationship between symptoms and exposure is often difficult to elicit. A high degree of clinical suspicion is required to confirm the diagnosis and initiate appropriate management. In a case series from Japan, the most frequent symptoms in chronic hypersensitivity pneumonitis were cough and dyspnea. 97 Fever (low-grade) occurred in only half of the patients.
Hypersensitivity pneumonitis must be distinguished clinically from the organic dust toxic syndrome, which is characterized by flulike symptoms occurring 4–8 hours after heavy organic dust exposures. 106 This syndrome, perhaps endotoxin mediated, does not relate to a specific antigen, and the chest radiograph is usually said to be normal or near-normal, though there are to our knowledge no specific descriptions of the CT appearances. The finding of specific IgG precipitins in the serum of a patient with suspected hypersensitivity pneumonitis serves as a confirmation of exposure, but is neither sensitive nor specific for hypersensitivity pneumonitis, and the precipitins do not appear to be important in pathogenesis. Serum precipitins are found in 3–30% of asymptomatic farmers and in up to 85% of asymptomatic pigeon breeders.100.107. and 108. Physiologic impairment in HP may be predominantly restrictive, predominantly obstructive, or mixed. Lalancette et al. 109 found obstructive abnormalities in 42% of farmers after 6 years of follow-up. Others have confirmed the high prevalence of airways obstruction, even after accounting for the effects of smoking. 110
On bronchoalveolar lavage, lymphocytosis is characteristic, with an inverted CD4/CD8 ratio in acute and subacute cases. In a study of 14 patients, the degree of lymphocytosis on lavage correlated with severity score for ground-glass abnormality, while those with normal CD4/CD8 ratio had a higher fibrotic score. 111 Hypersensitivity pneumonitis is characterized histologically by cellular bronchiolitis, a lymphoplasmacytic interstitial infiltrate, and poorly formed non-necrotizing granulomas. 112 However, this distinctive triad is often incomplete, particularly in subacute or chronic disease. Additional features may include giant cells, peribronchiolar metaplasia, 113 and organizing pneumonia. A fibrotic pattern very similar to UIP114 or NSIP115 is frequently seen in chronic, fibrotic hypersensitivity pneumonitis. In addition, the classic granulomas are seen in only 60–70% of acute cases and in less than 50% of chronic cases.97. and 116. In a review of 26 patients with chronic bird fancier’s lung, an NSIP pattern was seen in 13 and a UIP pattern in 11. 117
Radiographic features
The radiographic findings of acute hypersensitivity pneumonitis include diffuse ground-glass opacification (Fig. 8.13) and a fine nodular or reticulonodular pattern, which may be diffuse or may show lower lung predominance. 100 Consolidation is almost never seen, 118 and should lead to consideration of alternative diagnoses such as cryptogenic organizing pneumonia. A nodular or reticulonodular pattern becomes more prominent in the subacute phase (Fig. 8.14). 119 In chronic hypersensitivity pneumonitis, fibrosis with upper or lower lobe retraction, reticular opacity, volume loss, and honeycombing may be seen (Fig. 8.15). 97
The prevalence of an abnormal chest radiograph in population-based studies of patients with hypersensitivity pneumonitis is only about 10%.120. and 121. This relatively low sensitivity is probably due to the lack of conspicuity of the typical features of hypersensitivity pneumonitis. The sensitivity of CT for detection of hypersensitivity pneumonitis is much better than plain radiographs, but less than that of biopsy. 120 In a study by Lacasse et al., 122 16 (8%) of 199 patients with proven hypersensitivity pneumonitis had normal findings on HRCT.
Computed tomography
A variety of CT patterns has been described in hypersensitivity pneumonitis (Box 8.3).123.124. and 125.
Box 8.3
• Poorly defined centrilobular nodules
• Ground-glass abnormality
• Lobular decreased attenuation or air trapping
• Occasional cystic airspace
• Reticular abnormality
• Honeycombing
The nodules of hypersensitivity pneumonitis are round, poorly defined, and less than 5 mm in diameter (Figs 8.14 and 8.16). 118 They are typically centrilobular126 and profuse throughout the lung but a mid- to lower lung zone predominance has been variably reported. 127 In contrast to the centrilobular nodules of silicosis and other inhalational diseases, they are usually of ground-glass attenuation rather than of soft tissue attenuation. Profuse nodules of this type can be diagnostic of hypersensitivity pneumonitis in the correct clinical context. Although the nodules are centrilobular, a tree-in-bud pattern indicative of cellular bronchiolitis is quite uncommon.126.127. and 128. The nodules usually regress with removal from exposure.126. and 129.
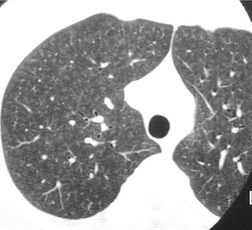
Ground-glass opacification in hypersensitivity pneumonitis (see Fig. 8.12) may be patchy or diffuse,125. and 127. and it may resolve with removal from exposure. 126 It is usually associated with restrictive physiology and impaired gas exchange on pulmonary function testing. 130 Ground-glass opacification is frequently found in association with other CT abnormalities such as centrilobular nodules, lobular decreased attenuation, or air-trapping (Fig. 8.12). Lung cysts may be seen in about 10% of cases of subacute hypersensitivity pneumonitis, but are usually sparse. 131 In chronic hypersensitivity pneumonitis, fibrosis is signified by irregular linear opacities, traction bronchiectasis, lobar volume loss, and honeycombing (Fig. 8.17).123. and 132. This abnormality may predominate in upper, mid- or lower lung zones, 127 but often spares the extreme lung bases.133. and 134. In the axial plane, the fibrotic changes are randomly distributed but a subpleural predominance has been described in some patients. 127 Conversely upper lobe fibrosis may occasionally be somewhat bronchocentric. Honeycombing (Fig. 8.18) may be found in up to 60% of cases.97.126. and 133. The presence of CT signs of fibrosis in patients with hypersensitivity pneumonitis correlates with histologic evidence of lung fibrosis, 132 and predicts poorer survival. Hanak et al. 135 followed up 26 patients with hypersensitivity pneumonitis who had CT signs of lung fibrosis and found that 11 had died, compared with 1 of 43 without evidence of fibrosis.
A mosaic attenuation pattern is common in hypersensitivity pneumonitis (Figs 8.12, 8.18 and 8.19), found in 80–90% of patients.130.133. and 136. In hypersensitivity pneumonitis, the mosaic pattern is usually due to a combination of patchy areas of ground-glass attenuation and lobular areas of decreased attenuation and vascularity due to air-trapping. When lobular decreased attenuation involves several lobules in more than four lobes, hypersensitivity pneumonitis is by far the most likely diagnosis. 133 Air-trapping on expiratory imaging is seen in many cases of hypersensitivity pneumonitis (Fig. 8.19), 136 and may be the predominant or only feature. HRCT evidence of air-trapping correlates with evidence of obstruction on pulmonary function testing.130. and 137. The air-trapping and mosaic attenuation are presumed to be due to bronchiolitis. 123
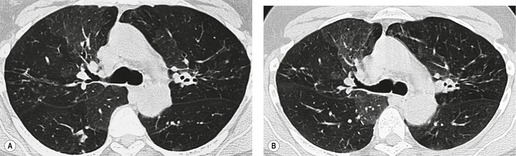
Fibrobullous lung destruction is (unlike in sarcoidosis) extremely unusual (Fig. 8.20). Radiographic evidence of emphysema in patients with chronic hypersensitivity pneumonitis was first noted by Barbee et al. 86 in 1968. Remy-Jardin et al. 126 noted emphysematous changes on HRCT in 50% of patients with chronic bird fancier’s hypersensitivity pneumonitis (Fig. 8.21). Both Lalancette et al. 109 and Cormier et al. 138 reported that emphysema occurred more commonly than fibrosis in chronic farmer’s lung. Another study showed an increased prevalence of emphysema in farmer’s lung patients compared with control farmers matched for age, sex, and smoking status. 124 In that study, 13 of the 20 farmer’s lung patients with emphysema were never smokers. Up to 50% of patients with hypersensitivity pneumonitis have mediastinal lymphadenopathy on CT, 139 but the enlarged nodes are almost always smaller than 2 cm in short axis. 139 Lymph node enlargement is therefore usually not visible on the chest radiograph.
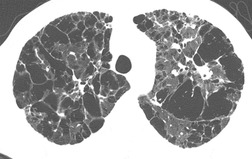
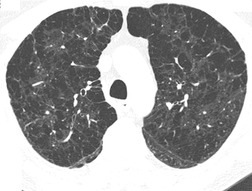
As with other types of fibrotic lung disease, acute exacerbations of chronic hypersensitivity pneumonitis may occur, and are characterized by extensive new ground-glass opacities, sometimes with focal areas of consolidation, superimposed on a fibrotic pattern. 140 The CT finding of consolidation is otherwise quite uncommon in hypersensitivity pneumonitis.
Relationship between imaging findings and phase of disease
The imaging findings in hypersensitivity pneumonitis do not necessarily correlate with the duration of symptoms. Centrilobular nodules, ground-glass attenuation, and mosaic attenuation may all be found at any phase of the disease, and may be the sole finding even in patients with clinically chronic hypersensitivity pneumonitis. Findings of fibrosis or emphysema are usually found only in chronic hypersensitivity pneumonitis.
Radiographic differential diagnosis
The differential diagnosis of hypersensitivity depends on the imaging findings. Profuse poorly defined nodules of ground-glass attenuation are virtually pathognomonic of hypersensitivity pneumonitis (Figs 8.14 and 8.16). The differential diagnosis may include smoking-related RB but in this condition the nodules are usually more patchy and sparse. Other diagnostic considerations should include pulmonary hemorrhage and metastatic pulmonary calcification. If the nodules are of soft tissue attenuation, sarcoidosis, chronic beryllium disease, or pneumoconiosis would be more likely than hypersensitivity pneumonitis. A patchy or unilateral distribution of nodules should suggest infection. 128
When ground-glass attenuation is present as the only sign of hypersensitivity pneumonitis, it may be indistinguishable from other conditions characterized by ground-glass attenuation such as viral infection, organizing pneumonia, DIP, or NSIP. Associated lobular hyperlucency should suggest the diagnosis of hypersensitivity pneumonitis, though the combination of ground-glass attenuation and hyperlucency may also be found in sarcoidosis.
Patients with chronic hypersensitivity pneumonitis may exhibit a histologic and imaging pattern of NSIP (Fig. 8.17) or UIP (Fig. 8.18). Hypersensitivity pneumonitis should always be considered in the differential diagnosis of NSIP115 and of UIP. 134 Imaging features which favor hypersensitivity pneumonitis over IPF or NSIP include lack of lower lung predominance, presence of centrilobular nodules, ground-glass abnormality or lobular air-trapping, and absence of honeycombing.133. and 134. However, these imaging features permit a confident distinction of chronic hypersensitivity pneumonitis from NSIP or UIP in only about 50% of cases. Therefore, hypersensitivity pneumonitis should always be considered in fibrotic lung disease, even when the imaging features are typical of NSIP or UIP.
Diagnosis (Box 8.4)
In a study of 400 patients (116 with hypersensitivity pneumonitis, and 284 controls with pulmonary symptoms), six significant clinical predictors of hypersensitivity pneumonitis were identified: exposure to a known offending antigen, positive precipitating antibodies to the offending antigen, recurrent episodes of symptoms, inspiratory crackles on physical examination, symptoms occurring 4–8 hours after exposure, and weight loss. 122 However, in many cases, particularly in chronic hypersensitivity pneumonitis, one or more of the characteristic features is absent. CT is often pivotal in suggesting the diagnosis. A history of exposure to a known antigen, or the presence of circulating precipitins, are inadequate evidence for the diagnosis of hypersensitivity pneumonitis. A recognizable exposure is absent in 25% of cases. 85 If the diagnosis remains equivocal after clinical and radiologic evaluation, bronchoscopy may help by demonstrating lymphocytosis on bronchoalveolar lavage or granulomas on transbronchial biopsy. Surgical lung biopsy is primarily used for cases in which there is no clear history of exposure, or where there is concern for other lung diseases.
Box 8.4
• Exposure to recognizable antigen
• Characteristic pulmonary/systemic signs and symptoms, often with latency after exposure
• Physiologic impairment (restrictive, obstructive, or mixed)
• Abnormal precipitins
• Characteristic imaging abnormalities (nodules, mosaic attenuation)
• Lymphocytosis on bronchoalveolar lavage
• Characteristic histologic abnormalities (granulomas, peribronchiolar infiltration)
*Not all are present in each case.
BYSSINOSIS
Byssinosis in cotton workers is characterized by respiratory symptoms which are most severe on the first day of the working week, and improve as the week progresses. Descriptions of the imaging findings of this entity have been sparse, but one report141 showed basal predominant ground-glass abnormality on the chest radiograph, with associated centrilobular nodules on CT.
PNEUMOCONIOSIS
The term ‘pneumoconiosis’, which means ‘dusty lungs’, is generally used to describe the non-neoplastic reactions of the lungs to inhaled dust particles. In general, the term excludes diseases due to organic dusts such as hypersensitivity pneumonitis or organic toxic dust syndrome. It also excludes asthma, bronchitis, and emphysema, which are common causes of symptoms and physiologic impairment in dust-exposed workers. The character and severity of the reaction of lung tissue to inhaled dust are determined by five basic factors:
• Properties of the inhaled dust, particularly the particle size and the degree to which the dust is fibrogenic
• Amount of dust retained in the lungs (respirable particles measuring between 0.5 µm and 5 µm are usually most likely to be retained, but asbestos fibers and talc particles may be larger than this)
• Duration and intensity of exposure
• Interval since the onset of exposure. A long latency from first exposure (20–30 years) is characteristic of pneumoconioses
• Individual idiosyncrasy.
The pneumoconioses are best classified according to the mineral dust responsible (Box 8.5). This classification is not always easy because industrial exposure may involve more than one dust known to be fibrogenic. For example, silica is so plentiful that some exposure to silica is inevitable in many forms of mining. Also, workers may be exposed to more than one type of dust during their career.
Box 8.5
• Due to dusts which are usually inert
– Iron – siderosis
– Tin – stannosis
– Barium – baritosis
• Due to dusts which cause interstitial fibrosis
– Beryllium – berylliosis
– Aluminum – aluminosis (Shaver’s disease)
– Asbestos – asbestosis
• Due to dust causing nodules. May progress to PMF
– Coal – anthracosis, coal worker’s pneumoconiosis
– Silica – silicosis
– Kaolin – kaolinosis
– Graphite – graphitosis
Radiographic classification of the pneumoconioses
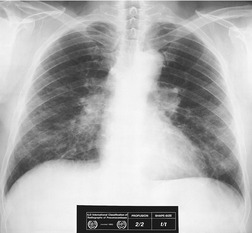
Occupational physicians have long recognized the need for quantitative techniques for evaluation of occupational lung diseases. This led to the development of the International Labor Organization (ILO) classification scheme for chest radiographs.142.143. and 144. In this system, the size, shape, and profusion of opacities on radiographs of patients with pneumoconiosis are classified in a detailed fashion by trained observers using a set of standard radiographs (Fig. 8.22). Regular (round) opacities are graded as p (less than 1.5 mm diameter), q (1.5–3 mm), or r (3–10 mm). Irregular opacities are classified as s, t, or u, using the same size criteria. Larger opacities are classified according to their size. The system also scores the extent and thickness of pleural plaques and pleural thickening, and provides symbols for other abnormalities such as fissural thickening and calcified nodules. There is now an international initiative to develop a similar standardized scoring system for CT.
Silicosis
Inhalation of free silica (silicon dioxide) can cause a variety of clinical abnormalities (Box 8.6). Free silica is present in many rocks in the Earth’s crust. It occurs in amorphous and crystalline forms, with quartz being the most important crystal. Exposed individuals usually work in quarries, drill or tunnel quartz-containing rocks, cut or polish masonry, clean boilers or castings in iron and steel foundries (fettlers), or are exposed to sandblasting. Although the incidence of silicosis has fallen since its peak during the second world war, it is still a major cause of pulmonary impairment in at-risk workers. The continued incidence of silicosis may be due to inadequacy of current respirable dust limits, or to failure to use appropriate respiratory protection.
Box 8.6
• Simple silicosis
– Diffuse upper zone predominant nodules, centrilobular on CT
– Calcification in pulmonary nodules and hilar and mediastinal nodes (often eggshell)
• Progressive massive fibrosis (PMF)
– Conglomeration of nodules in the posterior upper lobes
– Peripheral cicatricial emphysema
• Acute silicoproteinosis
• Chronic bronchitis
• Emphysema
• Tuberculosis and atypical mycobacterial infection
• Lung cancer
• Scleroderma
• UIP
• Pleural effusion, pleural thickening
In addition to the classic causes of silicosis outlined above, silicosis may also occur in ceramic145 or construction workers. 146 In Turkey, a newly recognized cause of accelerated silicosis and silicoproteinosis is sandblasting denim clothing.147. and 148. In a study of former denim sandblasters, chest radiographic evidence of silicosis was found in 77 of 145 subjects. 149 Other newly recognized occupational risk groups include goldworking jewelers150 and electric cable manufacturers. 151
The clinical presentation of silicosis may be acute, accelerated, or chronic, depending on the amount of dust exposure. The usual chronic form of the disease requires exposure to high dust concentrations for 20 years or more before radiographic abnormality is visible. 152 With very high concentrations of dust, radiographic changes may be visible in 4–10 years (accelerated silicosis).152. and 153. Acute silicoproteinosis is a clinically distinct entity that occurs with severe exposure, particularly in enclosed spaces. Patients with simple silicosis may be asymptomatic, or may have symptoms of chronic bronchitis. Symptoms are more common in those with greater profusion of disease. Most patients with complicated pneumoconiosis (PMF) have shortness of breath. Patients with acute or accelerated silicosis generally have marked respiratory impairment.
Pathogenesis and pathology of silicosis
Silica dust particles of appropriately small size are deposited on the alveolar walls and ingested by macrophages. They may enter the pulmonary lymphatics, or they may be transported to bronchioles for mucociliary clearance. The silica particle is directly cytotoxic to the macrophage. The dying macrophages induce fibrosis by releasing oxidants, proinflammatory cytokines, and stimulants of fibroblast proliferation. 154
The basic histologic lesion of silicosis is the hyalinized nodule. These nodules are usually more prominent in the upper zones and lie close to the bronchioles, small vessels, and lymphatics. They consist of concentric layers of collagen containing silica particles surrounded by a fibrous capsule. The outer zone is made up of irregularly dispersed connective tissue that also contains crystalline silica. The host response to this peripheral silica results in enlargement and conglomeration of nodules. The fibrosis seen with silicosis is both more abundant and more collagenous than that seen with other mineral dusts, such as coal. Silica-laden macrophages that reach the hilar and mediastinal nodes form granuloma-like lesions in the nodes.
Silicosis is often complicated by massive fibrotic lesions that are the result of the conglomeration of nodules matted together by fibrosis. These masses, which contain obliterated blood vessels and bronchi, may cavitate. The pulmonary macrophage, important in the granulomatous response to the infection, is damaged by silica particles rendering the silicotic individual more susceptible to mycobacterial or fungal infection. 155
Simple silicosis
The earliest radiographic changes of silicosis are 1–3 mm round, well-defined nodules, especially in the posterior portions of the upper two-thirds of the lungs (Fig. 8.23).156. and 157. A lower lung predominant reticular pattern may also be seen, either alone or in combination with the nodules. As the process advances, the nodules increase in size and number and become more widespread, involving all zones. Usually the nodulation is symmetric. Sometimes the nodules are calcified.
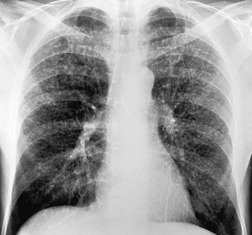 |
| Fig. 8.23 |
On CT scanning, the micronodules are usually centrilobular and perilymphatic (Fig. 8.24).158.159. and 160. As the disease evolves, there is spread anteriorly and inferiorly, although the upper zone preponderance is usually maintained. The upper lung preponderance is thought to be due to poor lymphatic clearance from the upper lobes. 161 Subpleural nodules may cluster to form ‘pseudoplaques’ (Fig. 8.25). 162 Larger nodules may also be seen, and may calcify. Interlobular septal thickening may be present. Not surprisingly, CT is clearly more sensitive than the chest radiograph for detection of nodules and of early conglomerate fibrosis. In a study of 44 silica exposed workers, CT identified small nodules in four workers who did not have nodules visible on chest radiograph, and identified PMF in 33 workers, compared with only 23 on chest radiograph. 163 CT scores for nodules and emphysema correlate with physiologic impairment.163. and 164. Interobserver agreement for the presence of nodules is substantially higher for CT than for chest radiograph (kappa value 0.84 compared with 0.54). 160 CT can objectively quantify the extent of emphysema and of lung nodules, and yields significant correlations with the level of physiologic impairment. 164
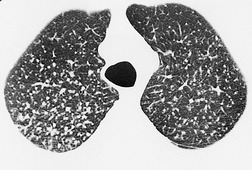 |
| Fig. 8.24 |
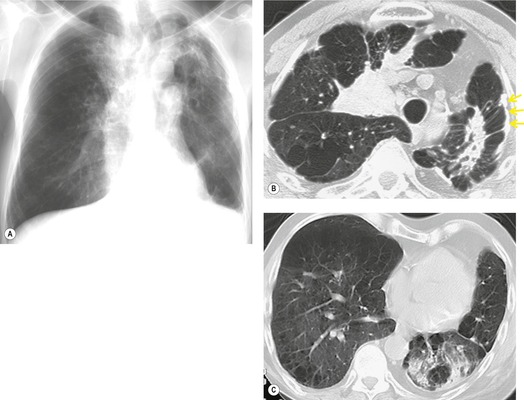
The obstructive component of physiologic impairment in silicosis has usually been attributed to emphysema. However, a study using paired inspiratory and expiratory CT in 34 workers with silicosis showed that air-trapping was in fact the most common finding in these individuals, and correlated quite strongly with a decreased forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio. 165 This study suggests that expiratory CT should be part of the routine CT evaluation of silica-exposed workers.
Hilar and mediastinal lymph node enlargement is quite common in silicosis. Calcification, sometimes of the eggshell type, may be seen in the nodes, either on chest radiograph (Fig. 8.26) or on CT. 166 In a study of 41 subjects with silicosis, lymph node classification was most commonly uniform or speckled; central, eccentric, or eggshell calcification was quite uncommon. The measured attenuation of lymph nodes correlated with the presence of PMF, with CT profusion of nodules, and with reduced lung diffusing capacity. 167
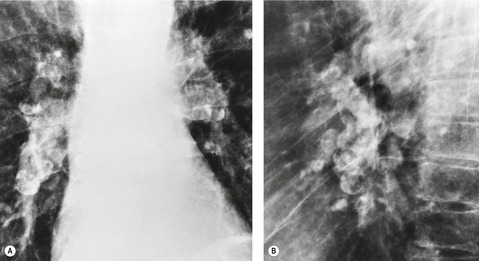
Progressive massive fibrosis
PMF may occur in silicosis or coal worker’s pneumoconiosis. Synonyms include complicated pneumoconiosis or conglomerate pneumoconiosis. This entity is defined radiographically by the presence of nodules greater than 1 cm in diameter. Recognition of PMF is important because it indicates that the patient is likely to be symptomatic and physiologically impaired. Typically, PMF starts as a round or oval mass near the periphery of the lung (Fig. 8.25), often with a well-defined lateral border that parallels the lateral chest wall. Lateral view confirms the posterior location and the oval appearance of the conglomerate mass (Fig. 8.27).
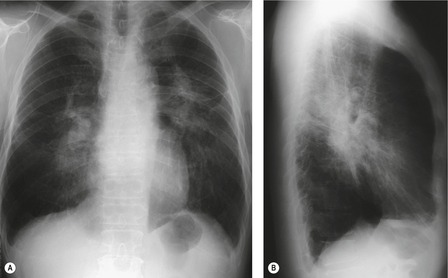
Masses of PMF, as the name implies, enlarge with time. The rate of growth is very slow, usually occurring over years. As the nodules coalesce and the upper lobes contract, the hila retract and compensatory emphysema occurs in the lower lobes. Apical pleural thickening is frequently seen. Some PMF lesions migrate medially toward the hilum, leaving a rim of peripheral cicatricial emphysema (Fig. 8.28). The migration is slow, taking 10 or more years to reach the hilum. Cavitation may be visible, and the cavities may empty and fill over a period of time. 168 Noncavitary large PMF lesions often show irregular low-attenuation regions on CT indicative of avascular necrosis. The presence of cavitation should always raise the suspicion of mycobacterial (including nontuberculous tuberculosis) superinfection. In 44 patients with silicosis and PMF studied by CT, 89% were located in the superior and posterior thirds of the lung. 169 Air bronchograms were seen within the masses in 70%, and calcifications in 64%. A study of 25 Brazilian sandblasters with PMF yielded very similar findings. 170
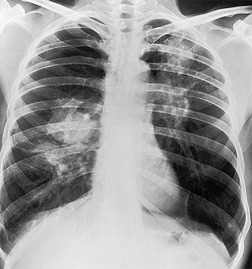
Since PMF is frequently bilateral, relatively symmetric, and accompanied by widespread nodules in the remainder of the lungs (Fig. 8.29), the diagnosis is rarely in doubt. There are cases, however, where the mass is completely or predominantly unilateral (Fig. 8.30) and background nodules may be minimal or absent. In such cases the differential diagnosis from lung carcinoma becomes particularly important. Features that favor the diagnosis of PMF include an indolent growth rate, oval or lenticular shape, location in an upper lobe close to and parallel to the major fissure, well-defined outer margin paralleling the chest wall, small irregular calcifications, upper lobe volume loss, and peripheral emphysema. 18-Fluorodeoxyglucose on PET-CT is of little assistance in resolving the differential diagnosis between lung cancer and silicosis, because increased uptake of this agent may be seen in lesions of PMF and in associated mediastinal lymph nodes (Fig. 8.31).171.172. and 173. Chong et al. 173 suggested that the presence of high signal on T2-weighted magnetic resonance imaging (MRI), and peripheral enhancement following gadolinium contrast administration, may be helpful in distinguishing lung cancer from PMF.
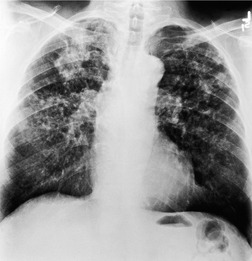
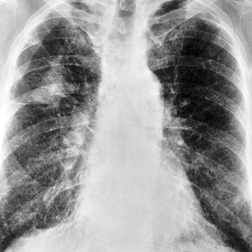
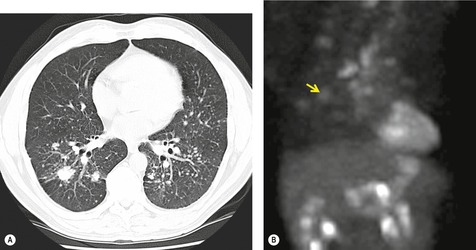
In addition to lung cancer, sarcoidosis must be considered in the imaging differential diagnosis of PMF, as the radiographic and CT features of lymphadenopathy, predominantly perilymphatic nodules and conglomerate fibrosis, in this condition may be very similar.
Other complications of silica exposure
While chronic simple silicosis refers to the development of silicosis between 10 and 30 years after exposure, accelerated silicosis refers to the development of silicosis within 10 years of exposure (usually related to more intense exposure). 174 The radiographic appearance of this entity is similar to that of chronic simple silicosis. However, workers with accelerated silicosis are at high risk for the development of complications such as PMF. 175 Intense exposure to silica dust, which may occur in sandblasters, may result in lung damage after only weeks or months, called acute silicosis or silicoproteinosis. The dominant histologic feature is the presence of an alveolar proteinaceous exudate, similar to that found in pulmonary alveolar proteinosis, hence the term ‘acute silicoproteinosis’.176. and 177. Death may ensue within 1–3 years.
On radiographic examination of patients with silicoproteinosis there is widespread alveolar opacity that progresses over a period of months. 178 The opacity is quite symmetric, but central and upper zone predominance is frequent (Fig. 8.32). Air bronchograms may be noted, and contraction of the lungs may be minimal at first, suggesting consolidation of the lungs. Hilar and mediastinal adenopathy may be seen, but the hila are often obscured by the parenchymal opacities. Peripheral air-trapping, bulla formation, lung volume loss, and distortion of mediastinal structures all indicate increasing fibrosis. Pneumothorax may occur. Silicoproteinosis may present with recurrent episodes of apparent pneumonia. 179
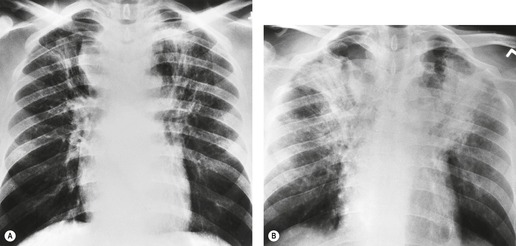
In a recent study of 13 patients (all sandblasters) with silicoproteinosis, the workers had been exposed for a median of 3.2 years, and presented with rapidly progressive respiratory symptoms. 180 Bilateral airspace consolidation was the most frequent finding on CT, found in 12 of 13 patients, and usually without zonal predominance. Foci of calcification were found within the areas of consolidation in 10 patients: this finding is unusual in other causes of consolidation and should strongly suggest the diagnosis. In the only patient without consolidation, the CT showed diffuse ground-glass abnormality and small centrilobular nodules. Centrilobular nodules were seen in 11 of 13 patients, and were usually diffuse. Confluent nodules were present in nine cases. Ground-glass opacity was seen in eight patients, but the crazy-paving pattern typical of pulmonary alveolar proteinosis was not identified in any case. However, a crazy-paving pattern was illustrated in some other reported cases.141. and 173. Calcified lymph nodes were present in 11 patients. Mild pleural thickening or pleural effusion was seen in 11 patients. Tracheal dilatation was present nine cases.
The relative risk of lung cancer in silica-exposed individuals ranges from 1.3 to 6.9, depending on the population, 179 but some of this excess risk is accounted for by cigarette smoking. Analysis is also complicated by the carcinogenic effect of radon gas exposure in miners. Nevertheless, both the American Thoracic Society179 and the International Agency for Research on Cancer181 have issued statements indicating that silica is a probable carcinogen and cause of lung cancer. It has not been established whether the risk is related to silica itself or to the pulmonary fibrosis induced by the silica.
Chronic bronchitis is probably the most common complication of silica exposure, often associated with airway obstruction. It has no radiographic manifestations, apart from airway wall thickening. Broncholithiasis with hemoptysis has been described in a patient with silicosis. 182 Silica exposure may be associated with emphysema (see Fig. 8.25). However, as with lung cancer, the confounding effect of cigarette smoking complicates analysis. Nonsmokers exposed to silica may develop emphysema, 183 particularly if they also have silicosis, 184 but the extent and physiologic significance of this emphysema are disputed. 185 It is clear, however, that, in patients with simple silicosis, the degree of impairment of pulmonary function is more closely related to the presence and severity of emphysema on CT than to the profusion of nodules.157. and 186. Most emphysema seen in association with silicosis is centrilobular, but cicatricial emphysema commonly occurs in association with PMF (Fig. 8.25).
Inhalation of silica predisposes to mycobacterial infection, classically with Mycobacterium tuberculosis, but infection with Mycobacterium kansasii, Mycobacterium avium complex, and other uncommon organisms has also been noted155. and 187. (Fig. 8.33). In one study, the relative risk of tuberculosis in workers with chronic silicosis was increased threefold compared with a control group. 179 The risk of mycobacterial infection is even greater in those with accelerated or acute silicosis, and in those with PMF. Mycobacterial infection in people with silicosis may be pulmonary or extrapulmonary. Pulmonary mycobacterial infection should always be suspected when cavitation, consolidation, or new nodules develop in a patient with silicosis (Fig. 8.33). 187
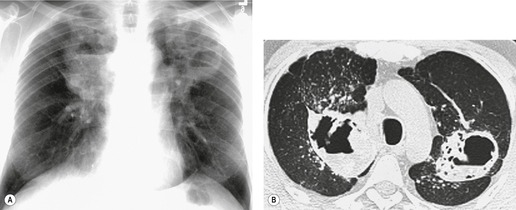
Silica workers have an increased risk of chronic interstitial pneumonia.188. and 189. The most common histologic pattern is UIP (Fig. 8.34). 190 Arakawa et al. 190 reviewed the CT scans of 243 workers with silicosis or mixed dust pneumoconiosis, and found a CT pattern of chronic interstitial pneumonia in 28 (11%). When compared with a group of patients with idiopathic pulmonary fibrosis, the overall extent of fibrosis and honeycombing was similar, but the extent of reticular and ground-glass abnormality, and of traction bronchiectasis, was less in those with pneumoconiosis, while the extent of nodules, emphysema, and subpleural homogeneous attenuation (shown histologically to represent confluent silicotic nodules) was greater. In a retrospective analysis of 14 patients with this form of chronic interstitial pneumonia, followed by CT for a median of 15.4 years, the extent and coarseness of fibrosis showed linear progression in 13 patients, and honeycombing developed in all patients over a median of 12.1 years. 191
Silica-exposed individuals are also at increased risk of scleroderma192. and 193. and perhaps of other collagen vascular diseases including rheumatoid arthritis, 179 dermatomyositis–polymyositis, 194 and systemic lupus erythematosus. 195 The prevalence of pleural abnormality in silicosis has been underemphasized: one report emphasizes that silicosis is associated with unexplained pleural effusions in 11% and pleural thickening in 58% of patients196 (Fig. 8.25). Rounded atelectasis may also be seen.197. and 198.
Coal worker’s pneumoconiosis (Box 8.7)
When an inhaled load of coal dust overwhelms the mucociliary clearance mechanism, dust-laden macrophages aggregate in the respiratory bronchioles and alveoli. The aggregation of dust and fibroblasts leads to the basic lesion of simple pneumoconiosis – the coal macule. If silica is released, collagenous fibrosis is stimulated. The coal macule develops around bronchioles, weakening the bronchiolar wall and leading to proximal acinar or focal emphysema.199. and 200.
Box 8.7
• Simple pneumoconiosis
• Progressive massive fibrosis (PMF)
• Chronic bronchitis
• Emphysema
• Diffuse interstitial fibrosis
• Lung cancer
Despite the similarity of their radiographic appearances, coal worker’s pneumoconiosis and silicosis are different pathologically. The coal macule does not have a hyaline center, nor does it show the laminated collagen typical of the silicotic nodule. The PMF lesion of coal worker’s pneumoconiosis is an ill-defined, amorphous, inhomogeneous collection of proteinaceous material, coal dust, and calcium phosphates. The factors governing the degree of lung damage caused by inhaling coal dust are not fully understood. Probably the most important factor is the amount of dust of suitable particle size that is inhaled. 201 The silica content (hardness) of the coal may also be important,201.202.203.204.205.206. and 207. but coal dust alone can cause pneumoconiosis and PMF.208. and 209.
Clinical features
It has been stated that simple coal worker’s pneumoconiosis is symptom free. 203 However, as with silica workers, coal miners commonly have symptoms of bronchitis and emphysema, features that are excluded from the definition of pneumoconiosis, but are responsible for significant symptoms and physiologic impairment.
PMF category A (nodules less than 5 cm) appears not to cause symptoms or signs. Categories B and C may be associated with respiratory disability and deterioration in ventilatory function tests. 203 Cough and sputum sometimes occur. Hemoptysis is rare. Cavitation of a PMF lesion may be associated with expectoration of jet-black sputum (melanoptysis). When this occurs, CT or MRI may show a fluid–fluid level within the cavity, and may show rim enhancement. 210
Radiographic appearances
The radiographic and CT signs of coal worker’s pneumoconiosis are usually indistinguishable from those described above for silicosis (Figs 8.29, 8.30, 8.35 and 8.36). Statistically, the main differences between coal worker’s pneumoconiosis and silicosis are that the nodules in coal worker’s pneumoconiosis tend to be smaller, and that silicosis is more likely to progress to PMF. PMF is uncommon in coal workers with less than 20 years’ experience in and around the mines. 211 Its appearance in coal workers is identical to that seen in silicosis (Fig. 8.27).
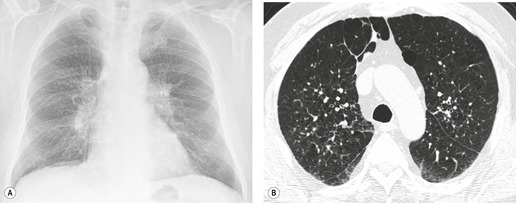
 |
| Fig. 8.36 (Courtesy of Dr. Martin Wastie, Nottingham, UK.) |
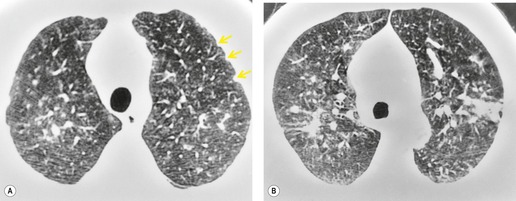
A minority (perhaps 10–20%) of coal miners develop diffuse lung fibrosis, characterized on the chest radiograph by small irregular opacities212. and 213. (Fig. 8.37). These irregular opacities correlate better than rounded opacities with the extent of physiologic impairment. 214 On CT, this entity is characterized by reticular abnormality often associated with honeycombing, similar to UIP or NSIP.215. and 216. This abnormality may or may not be associated with pneumoconiotic nodules. This pattern of diffuse interstitial fibrosis appears to be associated with a high prevalence of lung cancer, preferentially occurring in areas of lung fibrosis. 216 Other CT findings in coal worker’s pneumoconiosis may include bronchiectasis. 217
The chest radiograph reflects the extent and severity of the macules and masses found pathologically, and is therefore a good tool for diagnosing and following the progression of coal worker’s pneumoconiosis.156.168.202.218.219.220.221. and 222. The radiographic findings correlate well with the amount of dust to which the worker was exposed204 and the quantity of dust found in the lungs at postmortem examination. 223 However, the correlation between the chest radiograph and lung function is poor, except in patients with large conglomerate masses, 224 probably because the radiograph substantially underdiagnoses the associated emphysema.200. and 206. The degree of functional disability appears to correlate far better with the severity of emphysema than it does with the presence of macules or PMF.157.200.221. and 225. The etiologic role of smoking in coal worker’s emphysema is controversial. Some studies have shown that coal miners have a degree of emphysema that is excessive even if allowance is made for age and smoking history.226. and 227. This applies particularly to patients with PMF. At lesser degrees of pulmonary involvement, smoking may play a more substantial role in the causation of the emphysema and hence the patient’s symptoms.157. and 228.
Although the pace of progression of coal worker’s pneumoconiosis is usually slow, a subset of workers develop rapidly progressive pneumoconiosis, defined as development of new PMF or increase in profusion of small opacities by more than one subcategory under the ILO classification system in 5 years or less. A recent study found a total of 886 cases of coal worker’s pneumoconiosis among 29 521 workers evaluated between 1996 and 2002. 229 In a subset of 783 in whom progression could be evaluated, 277 (35%) had rapidly progressive pneumoconiosis. The radiologist should recognize the occurrence of rapidly progressive pneumoconiosis, as it is often a sentinel event, indicating inadequate preventive measures at the worksite. 229
Mixed dust pneumoconiosis
Mixed dust pneumoconiosis occurs with inhalation of a mixture of silica and nonfibrous silicates. 230 Typical occupations associated with the diagnosis of mixed dust pneumoconiosis include metal miners, quarry workers, foundry workers, pottery and ceramics workers, and stonemasons. Silica is much more fibrogenic than the silicates, so often the imaging appearances of silicosis predominate on pathology and imaging. More pure mixed dust pneumoconiosis is characterized histologically by dust macules and a diffuse interstitial fibrosis pattern, similar to idiopathic pulmonary fibrosis. On the chest radiograph and CT it is characterized by fine reticular abnormality.
Asbestos-related diseases
Asbestos comprises a group of fibrous silicates with differing chemical and physical properties but with the notable common property of heat resistance (Box 8.8). Chemical differences such as solubility and acid resistance, and physical differences such as fiber length, brittleness, and texture are probably important determinants of the distribution and severity of deleterious effects on the lungs and pleura. Asbestos is divided into two principal subgroups based on the physical properties of the fibers: the serpentines and the amphiboles. Serpentine asbestos has long, curly, flexible, smooth fibers composed of fibrillary subunits. The only serpentine asbestos used commercially is chrysotile, which accounts for over 90% of the asbestos used in the USA today. 231 The amphiboles have straight, needlelike fibers of varying length, diameter, brittleness, and texture. The main types in use are crocidolite (blue asbestos), amosite (brown asbestos), tremolite, and anthophyllite. The longer, less soluble amphibole fibers tend to be very slow to clear from the lung, compared with chrysotile. A major reason for the decline in the use of the amphiboles was the recognition that the amphiboles, particularly crocidolite, have a greater fibrogenic and carcinogenic potential than the serpentine-form chrysotile. 232 The carcinogenic risk of chryosotile is related to fiber length, but risk of asbestosis is less clearly related to fiber dimensions. 233
Box 8.8
A family of naturally occurring fibrous silicates highly resistant to physical or chemical destruction
Two fiber types
• Serpentine asbestos – long curly fibers composed of fibrillary subunits
– Chrysotile – most common, less fibrogenic and carcinogenic
• Amphibole asbestos – straight shorter needlelike fibers
– Crocidolite
– Amosite
– Anthophyllite
– Tremolite
Uses of asbestos
• Manufacture of cement, bricks, tiles, paints, fireproof felts, and textiles
• Manufacture of furnace and oven linings
• Insulation of steam pipes and electric lines
• Brake lining manufacture
• Fire proofing and insulation of buildings
World production and use of asbestos expanded to an extraordinary degree over the past century. Production expanded from 50 tons per annum in the 1860s to a peak of over 5 million tons per annum in the early 1970s. 234 Asbestos production has now declined somewhat as alternative materials have become available. Nevertheless, with the long latency which exists before the adverse effects of asbestos exposure become manifest (Box 8.9, Table 8.3), and with the vast tonnages of the material now in place or in use, asbestos is likely to remain one of the major environmental hazards well into the twenty-first century, particularly in the developing world.235.236. and 237. A recent outbreak of asbestos-related pleural disease and mesothelioma related to a vermiculite mine in Libby, Montana, USA, illustrates the need for continued vigilance for asbestos-related disease. The vermiculite was contaminated with small amounts of tremolite, which was sufficient to cause a considerable increase in mortality from lung cancer, mesothelioma, and asbestosis.238.239. and 240.
Box 8.9
• Benign asbestos related pleural effusion(s)
• Diffuse pleural thickening
• Pleural plaque formation
• Rounded atelectasis
• Asbestosis
• Malignancies
– Lung cancer and other epithelial malignancies – larynx, gastrointestinal tract
– Mesothelioma: pleural, peritoneal, pericardial
| *Adapted with permission from Lynch et al. Imaging of diffuse lung disease. Hamilton, Ontario: BC Decker 2000. | ||
| †The above figures are provided only as approximate guidelines. The prevalence or incidence of disease depends on the duration and intensity of exposure. | ||
| ‡Lifetime incidence in heavily exposed workers. | ||
| Finding | Latency (years) | Approximate frequency in asbestos workers (%) |
|---|---|---|
| Asbestos-related effusion | 5–20 | 3 |
| Noncalcified plaques | 15–30 | 15–80 |
| Calcified plaques | 30–40 | 10–50 |
| Diffuse pleural thickening | 10–40 | 10 |
| Asbestosis | 20–40 | 15–30 |
| Lung cancer | >15 | 20–40‡ |
| Mesothelioma | 15–40 | 10‡ |
Benign asbestos pleural effusion
Benign asbestos-related pleural effusion is one of the less common manifestations of asbestos exposure, but it has the shortest latency period241. and 242. (Box 8.10). Diagnosis of this condition is sometimes missed because of the transient, often asymptomatic nature of these effusions and the lack of specific markers to indicate their cause. Indeed the diagnosis is one of exclusion and should conform to the following criteria:
• History of occupational or environmental asbestos exposure
• No other cause for the effusion
• No evidence of malignancy within 3 years of detecting the effusion.
Box 8.10
• Benign asbestos-related pleural effusion(s)
– May be the first manifestation of asbestos exposure
– Unilateral or bilateral
– Often asymptomatic; otherwise fever, pleuritic chest pain, or dyspnea
– Usually small but may be >500 mL
– Fluid – exudative often blood tinged
– Usually resolve completely but may leave diffuse pleural thickening
• Diffuse pleural thickening.
– Usually follows effusion.
– Costophrenic sulcus blunted.
– Usually asymptomatic but may cause pulmonary restriction
• Pleural plaques.
– Usually located on parietal pleura.
– Most commonly seen between the fourth and eighth ribs, particularly under anterior ribs.
– No functional significance – they are a marker of asbestos exposure
• Malignant mesothelioma
In an epidemiologic study by Epler et al., 241 the overall incidence of benign pleural effusion was 3.1%, with a 7% incidence among individuals having a heavy occupational exposure and a 0.2% incidence in environmentally exposed individuals. Effusions may be unilateral or bilateral and tend to recur. The amount of fluid is usually small; effusions greater than 500 mL are uncommon. The fluid has the characteristics of an exudate and may be blood tinged. Symptoms include pleuritic pain, fever, and an elevated white blood cell count, but many patients have only mild symptoms or no symptoms at all. Indeed the presence of substantial chest pain should lead to concern for mesothelioma.
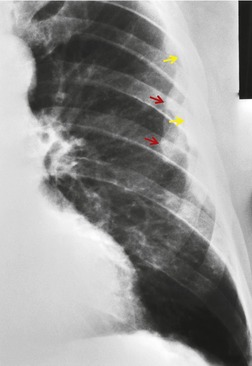
Benign pleural effusion is the most common abnormality seen within 10 years of the onset of asbestos exposure. However, it may occur up to 58 years after initial exposure. 243 No direct causal relationship between benign effusions and the subsequent development of malignant pleural effusion has been postulated, but in one study of 70 cases of malignant mesothelioma, 244 five cases were preceded by what were regarded as successive benign pleural effusions for up to 7 years. In asbestos workers with pleural effusions, the diagnosis of benign effusion should be a diagnosis of exclusion. Mesothelioma should be considered in any patient with a latency longer than 10 years, or in any patient with chest wall pain. In one series of 312 cases of mesothelioma, the latency periods ranged from 14 to 72 years with a mean of 48 years. 245 There appeared to be an inverse relationship between latency and the intensity of exposure; for example, the mean latency period for insulators was 30 years compared with 52 years for domestically exposed women. Benign pleural effusions are associated with the subsequent development of diffuse pleural thickening (Fig. 8.38).246. and 247. McLoud and co-workers248 found that just over 50% of their patients with benign pleural effusions subsequently developed diffuse basal pleural thickening, an association also emphasized by Cookson and associates. 249
Asbestos-related pleural plaques
Irregular pleural thickening and calcification were first positively related to exposure to asbestos in 1955, 250 and the occurrence of noncalcified, asbestos-induced plaques was first reported in 1967. 251 These changes represent the most frequent radiographic manifestation of exposure to asbestos. Although the incidence of pleural plaque formation increases with dose, the elapsed time from initial exposure is a more important factor. 252 Pleural plaques are usually first identified more than 20 or 30 years after the initial asbestos exposure. 253 Asbestos-induced pleural plaques occur on the parietal pleura, especially over the diaphragm and along the posterolateral chest wall. The apices, the costophrenic sulci, and the mediastinal pleura tend to be spared. The plaques are composed of elevated areas of hyalinized fibrous tissue and frequently lie beneath the ribs. Although usually isolated, they may enlarge, spread, and coalesce. Dystrophic calcification within plaques is common and is more frequent as the plaques age and enlarge. Electron microscopy studies have identified asbestos fibers in the plaques on the parietal pleura, indicating that transpleural migration and assimilation of inhaled asbestos fibers must occur. 254 There is evidence that the widely used, more benign form of asbestos, chrysotile, is particularly associated with this transpleural migration, while the more fibrogenic and carcinogenic amphiboles, crocidolite and amosite, tend to be retained in the lung parenchyma.253. and 255. This may account for the common finding of asbestos-related pleural disease unassociated with parenchymal fibrosis or intrathoracic malignancy.
Population surveys in industrial regions and in asbestos-mining areas have revealed a surprisingly high incidence of pleural plaques even in individuals who have only a remote connection with asbestos.256.257. and 258. Some of these plaques may be due to incidental asbestos exposure, but some ‘plaques’, detected on chest radiograph, may be due to extrapleural fat. In autopsy-based studies of unselected populations, pleural plaques may be found in up to 20% of patients, and a history of asbestos exposure is lacking in most of these,259. and 260. particularly where the plaques are small or unilateral. 260 Such plaques may be due to unrecognized asbestos exposure or to other pleural injury. Pleural plaque formation and calcification have been noted to occur after contact with other nonasbestos fibrous minerals such as erionite in Turkey261 and sepiolite in Bulgaria. 262
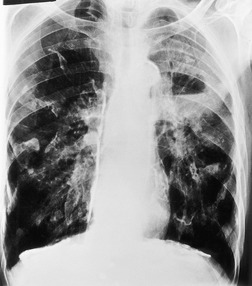
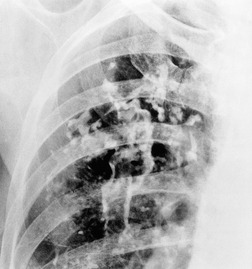
On chest radiographs, noncalcified pleural plaques are identified as smooth elevations of the pleura in profile along the chest wall (Fig. 8.39) or over the diaphragm. Plaques are difficult to identify en face unless they are large or calcified. En face plaques are seen as amorphous areas of increased density. They are relatively flat in relation to their width, and the density of the opacity projected over the lungs is therefore less than would be expected for a parenchymal lesion of equivalent size. Furthermore, one margin of the lesion is likely to be indistinct as it smooths off into normal pleura. Plaques are usually multiple and bilateral. Although Hu et al. 263 reported that pleural plaques detected on chest radiographs were statistically more common on the left, a left-sided predominance was not confirmed in a CT-based study. 264
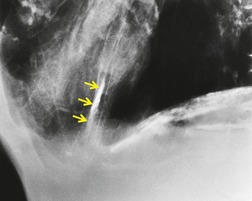
Calcification in pleural plaques is linear when seen in profile (Fig. 8.40), but when it is seen en face the appearance is variable (Fig. 8.41), the most common type being ‘holly leaf’ calcification (Fig. 8.42). Extensive pleural calcification may be lacelike. Pleural plaques may occur on the visceral pleura, including the interlobar fissures (Fig. 8.43).265. and 266. Smooth visceral pleural thickening due to plaque must be distinguished from the more irregular subpleural fibrosis which occurs in patients with asbestosis. 267 Asbestos exposure may be associated with pericardial fibrosis, with or without calcification or effusion.268. and 269. Yazicioglu, 270 in a study of 511 cases of pleural disease secondary to environmental exposure to chrysolite asbestos in Turkey, found 1.7% of cases with coincident pericardial calcification. Asbestos-related pericardial fibrosis may lead to constrictive pericarditis.268. and 271.
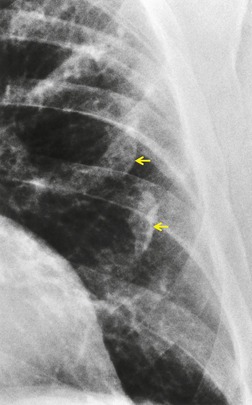
The differential diagnosis of pleural plaques includes extrapleural fat deposition in obesity, 272 extrapleural thickening in relation to multiple rib fractures, postinflammatory pleural thickening, and pleural metastases. In practice, the most frequent simulator of asbestos-related pleural plaque formation is extrapleural fat deposition. 273 Extrapleural fat may be differentiated from pleural plaques by its lower density, its smooth, undulating outline, and by the fact that it typically extends all the way to the lung apices (Fig. 8.44). However, this distinction is sometimes inaccurate. Fat is most abundant over the fourth to eighth ribs and does not involve the costophrenic sulci. CT will readily distinguish between soft tissue density of pleural plaques and extrapleural fat.
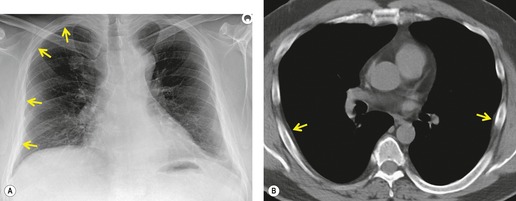
Although the presence of pleural plaques is associated with less physiologic impairment than diffuse pleural thickening, several studies have indicated that identification of plaques on chest radiographs is associated with evidence of pulmonary restriction, independent of the presence or extent of radiographic parenchymal abnormality.274.275.276.277. and 278. Some of this association might be explained by the presence of subradiographic asbestosis.279. and 280.
CT is substantially more sensitive than the chest radiograph for detection of pleural plaques (Fig. 8.45).281.282.283. and 284. Contiguous CT imaging maximizes sensitivity for plaques. 285 CT is also more sensitive than radiographs for detecting calcification in plaques. 286 Elevated plaques may indent the adjacent lung, and may even cause ground-glass abnormality by interfering with subjacent pulmonary expansion. Thinner plaques may be difficult to distinguish from the intercostal musculature, but may be identified by recognizing that they overlay ribs as well as intercostal spaces (Fig. 8.46). Meirelles et al. 287 developed a detailed semiquantitative scoring system for determining extent of pleural plaques, which might be of value in assessing correlation with physiology. Weber et al. 288 found that high-resolution and radial MRI was almost as sensitive as CT for detection of pleural plaque. However, since CT is more sensitive for parenchymal abnormalities, it seems likely that it will remain the primary technique for detection of asbestos-related pleural disease.
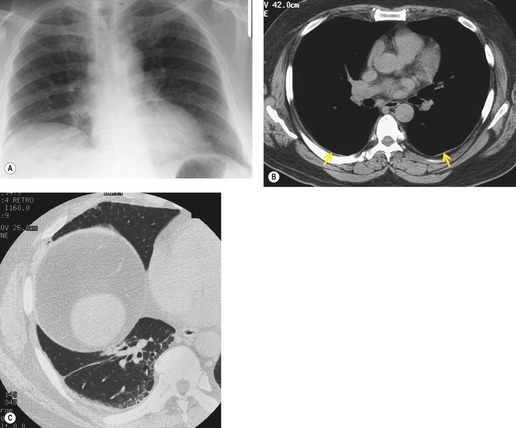
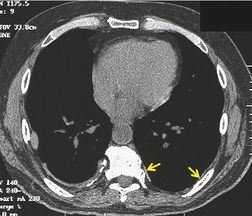
Diffuse pleural thickening
It is important to distinguish between asbestos-related diffuse pleural thickening and pleural plaques because patients with diffuse pleural thickening commonly have marked impairment of pulmonary function. The radiographic definition of diffuse pleural thickening has varied. The ILO classification defines it as pleural thickening that involves the costophrenic sulci (Fig 8.47), 143 while McLoud et al. 248 also included patients in whom pleural plaques occupied more than a quarter of the chest wall. Most studies have used the more restrictive ILO definition. On CT scanning, the definition of diffuse pleural thickening has also varied. Lynch et al. 267 defined it as an area of pleural thickening more than 3 mm thick, more than 5 cm in transverse dimension, and more than 8 cm in craniocaudal dimension. However, Copley et al. 289 defined diffuse pleural thickening as pleural thickening with tapered margins, in contrast to pleural plaques, which were required by this definition to have well-defined margins. Since diffuse pleural thickening, by all definitions, is associated with physiologic impairment,277.289. and 290. the precise definition of this entity is of little importance.
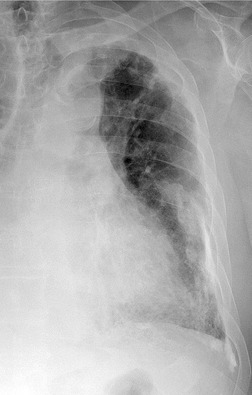
Asbestos-related diffuse pleural thickening must be distinguished from the visceral pleural and subpleural fibrosis which occurs in patients with many forms of lung fibrosis (including asbestosis). Pure parietal pleural thickening is usually sharply defined on CT, while visceral subpleural fibrosis is associated with fine fibrous strands extending into the underlying lung, giving an irregular demarcation to the pleural process (Fig. 8.48).267. and 291. Visceral subpleural fibrosis is usually, but not always, associated with other evidence of lung fibrosis. Differential diagnosis of asbestos-related diffuse pleural thickening includes pleural thickening (usually unilateral) related to prior empyema, infection, or other cause. Interestingly, patients with asbestos-related diffuse pleural thickening have been found to have a more frequent history of coronary bypass surgery than other asbestos-exposed individuals, suggesting that thoracic surgery may have a synergistic role in development of this complication. 290 Diffuse pleural thickening can usually be differentiated from mesothelioma by the absence of pleural effusion or pleural masses.
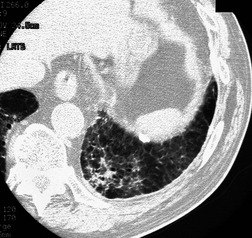
Rounded atelectasis
Rounded atelectasis, also called folded lung, 292 is a masslike area of lung collapse adjacent to an area of pleural thickening. The condition is not unique to asbestos-related pleural thickening, and may also be seen with any other cause of exudative or organizing pleural disease. 293 Though typically associated with benign pleural disease, it may sometimes be seen with mesothelioma. 294 Hanke and Kretzschmar295 proposed that rounded atelectasis occurs when an atelectatic area of lung becomes infolded during resorption of a pleural effusion. Other authors,296. and 297. noting that rounded atelectasis is not always preceded by a pleural effusion, have suggested that it may result from centripetal contraction of a focus of visceral pleural fibrosis, causing buckling of the pleura and collapse of the underlying lung parenchyma.
The radiographic findings are characteristic (Box 8.11) (Fig. 8.49




Related posts:
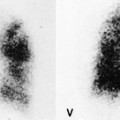
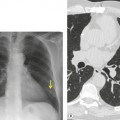
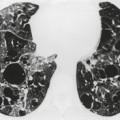
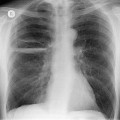
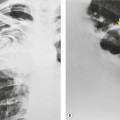
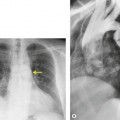
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
