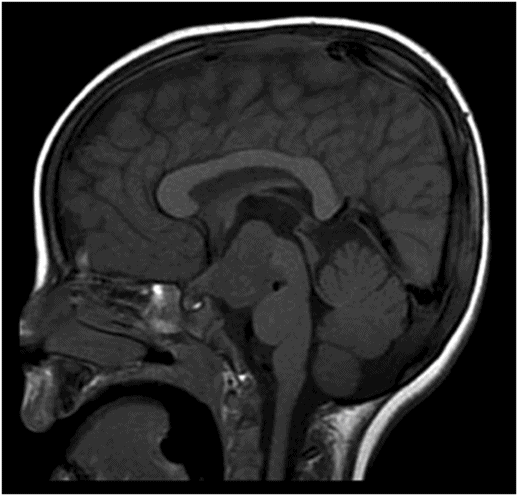
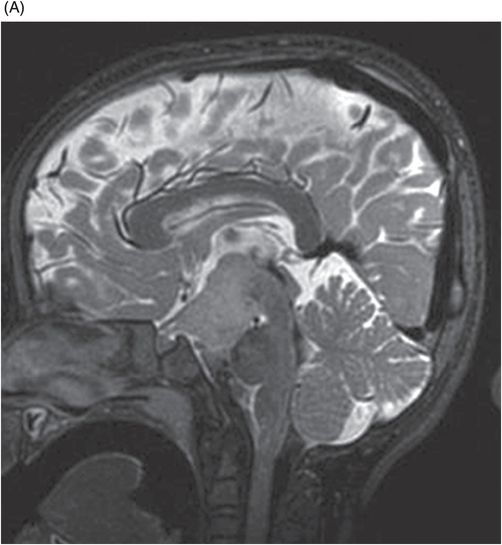
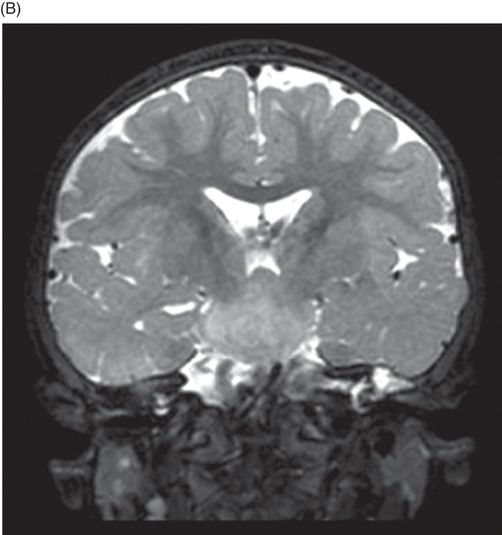
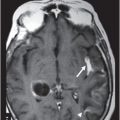
(A) Midsagittal T2WI of the brain through corpus callosum and hypothalamo-pituitary axis. (B) Coronal T2WI of the brain through the pituitary and suprasellar region at the level of anterior body of lateral ventricles.
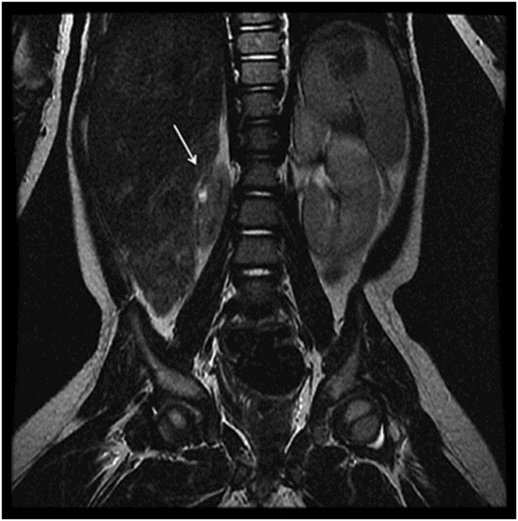
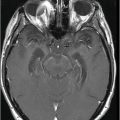
Coronal T2WI of the abdomen through the kidneys.
Pallister-Hall Syndrome
Primary Diagnosis
Pallister-Hall syndrome
Differential Diagnoses
Isolated hypothalamic hamartoma
Craniopharyngioma
Hypothalamic-opticochiasmatic glioma
Complex syndromic associations
McKusick-Kaufman syndrome
Smith-Lemli-Opitz syndrome
Oral-facial-digital syndrome type VI
Greig cephalopolysyndactyly syndrome
Imaging Findings
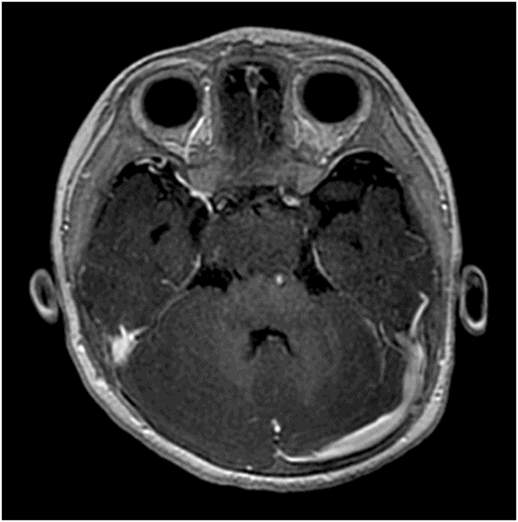
Fig. 119.2: Midsagittal T1WI and Fig. 119.3: (A) Midsagittal T2WI through the corpus callosum and hypothalamo-pituitary axis, and Fig 119.3: (B) Coronal T2WI through the pituitary and suprasellar region at the level of anterior body of lateral ventricles showed a large hypothalamic mass lesion following gray matter signal characteristics. Fig. 119.4: Axial T1W postgadolinium image through the mass lesion did not show any enhancement. Fig. 119.5: Coronal T2WI of the abdomen shows a hypoplastic right kidney with a small cyst (arrow).
Discussion
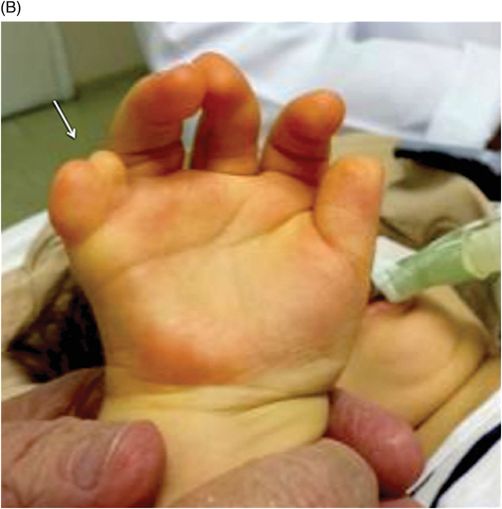
The constellation of clinicoradiologic findings comprising polydactyly, anomalies involving the upper aerodigestive tract and genitourinary system, accompanied by hypothalamic hamartoma confirms the diagnosis of Pallister-Hall syndrome. On MR imaging, hamartomas have signal characteristics resembling gray matter with no evidence of calcification or enhancement. Although very rare, cystic degeneration and liquefactive necrosis changes have been described in the literature.
Craniopharyngiomas have a more heterogeneous solid-cystic pattern and tend to have areas of calcifications, while hypothalamic-opticochiasmatic gliomas have predominantly T2 hyperintense signal and variable enhancement.
There is a considerable degree of multisystem involvement among the above-mentioned list of syndromic differential diagnoses, but the presence of hypothalamic hamartoma is the key feature of Pallister-Hall syndrome. Hypothalamic hamartoma can also be associated with oral-facial-digital syndrome type VI; however, it has other features resembling Joubert syndrome-related disorders with posterior fossa malformations that are not seen with Pallister-Hall syndrome. Mutations in GLI3 gene can cause Greig cephalopolysyndactyly syndrome. Macrocephaly, peculiar skull shape due to craniosynostosis, callosal agenesis, and mental retardation are some of the imaging and clinical features apart from the other overlapping multisystem findings. McKusick-Kaufman and Smith-Lemli-Opitz syndromes share common non-CNS multisystem involvement but hypothalamic hamartoma association is rare.
Judith Hall first described Pallister-Hall syndrome in 1980. It is a rare autosomal dominant pleiotropic disorder caused by GLI3 gene mutation on chromosome 7p13. As only a few cases have been described its exact prevalence is unknown. The Pallister-Hall spectrum consists of polydactyly, renal and pulmonary segmentation anomalies, imperforate anus, dysplastic nails, bifid epiglottis, panhypopituitarism, and hypothalamic hamartoma. Other craniofacial associations include cleft palate, cleft uvula, cleft larynx, buccal frenula, flat nasal bridge with short nose, and low set, posteriorly angulated ears. Although the pituitary manifestations can present with acute adrenal insufficiency or hypothyroidism, they can also be asymptomatic.
On histologic examination, a hamartoma consists of a disordered arrangement of neurons mixed with astrocytes and white matter, originally described as hypothalamic hamartoblastomas.
It is important to diagnose isolated hypothalamic hamartomas, as affected patients are young and present with early-onset seizures that are resistant to antiepileptic drugs. In addition, these individuals may present with precocious puberty and cognitive impairment. Surgical intervention for hamartoma is associated with poor outcome. In contrast, patients with Pallister-Hall syndrome-associated hamartoma presenting with gelastic seizures respond well to antiepileptic medications. The extent of behavioral and cognitive disabilities is also less severe than isolated hypothalamic hamartoma. Recognizing this entity is extremely important in order to offer patients and their families genetic counseling and screening, and to correlate any associated endocrinologic symptoms.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


