It is apparent from clinical experience that congenital or developmental lesions of the lungs are neither as frequent nor, in general, as significant as their counterparts in the heart. Evidence from autopsy series supports this impression. For example, Sotelo-Avila and Shanklin1 showed that some form of congenital malformation occurred in 46% of over 2000 autopsies. Of these, 30% were cardiovascular and 10% were genitourinary, but only 3% were respiratory in nature.
Classification of congenital lesions of the lungs and airways is difficult because the embryogenesis of specific pulmonary malformations is often obscure. Although some of the conditions are not present at birth, they are nevertheless developmental in origin. Because the lungs continue to form for an extended time after birth, deleterious factors operating post natally can affect their development as, for example, in Swyer–James syndrome (see p. 750). Classification is further complicated by associated congenital heart disease, which can lead to difficulty in deciding whether a particular malformation should be categorized as a lung lesion or as a cardiovascular anomaly.2. and 3.
Box 16.1 shows just one of many arbitrary classifications of developmental lesions of the thorax. In relation, many of the more common conditions are discussed in this or other chapters; the less common or frankly rare conditions are only referenced.
Box 16.1
Abnormalities of development, branching, or separation of the foregut
• Bronchopulmonary foregut malformations9
• Bronchial isomerism syndromes10
• Congenital lobar overinflation
• Ectopia of the lungs11
• Horseshoe lung
• Hypoplasia of the lungs
• Potter syndrome (oligohydramnios tetrad)
• Tracheal diverticulum14
• Tracheal agenesis15
• Tracheal stenosis16
• Tracheal abnormalities in skeletal dysplasias17
• Tracheomalacia and tracheobronchomegaly (Mounier–Kuhn syndrome)
• Tracheoesophageal fistula (16.4)
Abnormalities related to a local or systemic biochemical or cellular defect
Abnormalities of the pulmonary vasculature or lymphatics
Abnormalities forming a component of a systemic familial or nonfamilial disease process
• Hereditary hemorrhagic telangiectasia
• Pulmonary lymphangioleiomyomatosis (see p. 685)
• Potter syndrome (oligohydramnios tetrad)
Familial abnormalities of the lungs
TRACHEOESOPHAGEAL FISTULA
Most tracheoesophageal fistulas are associated with esophageal atresia and are diagnosed in the neonatal period. However, in rare instances, the so-called H-type fistula may not be detected until adult life even though symptoms may have been present from infancy28 (see later Fig. 16.4). The H-type fistula connects the adjacent trachea and esophagus, with the fistula forming the transverse bar of the ‘H’. This type of fistula constitutes approximately 2–4% of all tracheoesophageal fistulas.29. and 30. There is a relatively high association between tracheoesophageal fistula and other congenital abnormalities. 31 These associated anomalies can provide a clue to the presence of an H-type fistula. An extensive literature describes the differing patterns of associated anomalies. 10 These patterns have been designated by a series of acronyms: the VATER complex (vertebral, anal, tracheoesophageal, and renal), the VACTEL complex (vertebral, anal, cardiac, tracheoesophageal, and limb), and the ARTICLE ± V complex (anal, renal, tracheal, intestinal, cardiac, limb, and esophageal ± vertebral). Other congenital pulmonary lesions such as pulmonary hypoplasia, tracheal stenosis, and pulmonary sequestration occur in approximately 2% of patients with this group of complex malformations. 32
Symptoms of congenital tracheoesophageal fistula are generally due to recurrent aspiration and include paroxysmal cough, feeding difficulties, and recurrent pneumonia. Pulmonary opacities seen on chest radiographs reflect the extent and severity of aspiration. Excessive air may pass through the fistula into the esophagus and subsequently into the gut. In infants with an H-type fistula, an air-filled esophagus may be seen on the chest radiograph and there may be unusual degrees of gaseous distension of the bowel. 33 The condition can be difficult to diagnose with contrast studies of the esophagus because the fistula may be relatively small and aspiration through it may be intermittent. 34 The patient should be examined in the prone position, with lateral fluoroscopy and video recording. It is advantageous to inject the contrast medium through a feeding tube while it is withdrawn along the full length of the esophagus. In problematic cases, thin barium suspensions or nonionic water-soluble contrast media are useful. The purpose of these measures is to encourage the contrast medium to enter the fistula. 35 In some cases, repeating the contrast studies on more than one occasion may be necessary to establish the diagnosis. 36 Thus, diagnosis clearly requires a high degree of suspicion.
BRONCHIAL ATRESIA
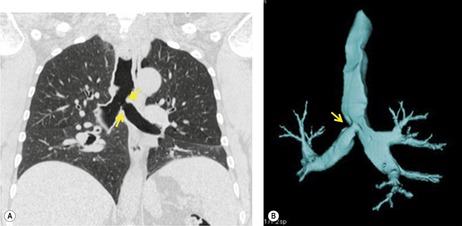
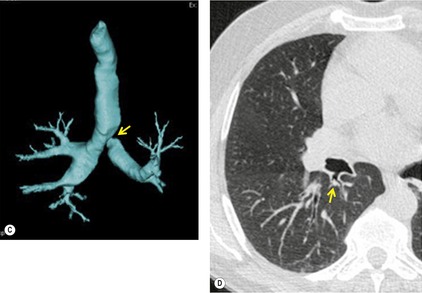
Segmental bronchial atresia (Box 16.2) is a rare anomaly that results from short-segment obliteration of a proximal segmental or subsegmental bronchus.37. and 38. The distal airways and airspaces develop normally, producing the characteristic triad of findings:39. and 40.
Box 16.2
Etiology
• In utero vascular accident (probable)
• Proximal short-segment atresia of segmental bronchus
• Distal airway patent
• Distal lung aerates via collateral pathways
Symptoms
• Most asymptomatic
Location
• Upper lobes most common
• Usually apical or posterior segments
Chest radiography
• Central masslike opacity (mucocele)
• Surrounding hyperlucent lung
Chest CT
• Central masslike opacity (mucocele)
• Surrounding hyperlucent lung
• Used to confirm radiographic findings
• Used to exclude endobronchial mass (tumor)
• Air-trapping on expiratory images
Differential diagnosis
• Allergic bronchopulmonary aspergillosis
• Endobronchial mass (tumor, foreign body)
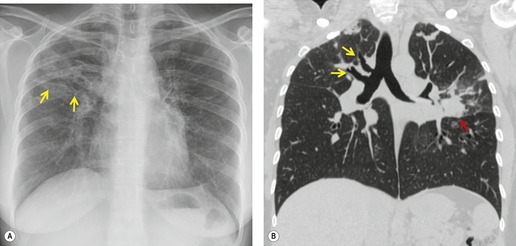
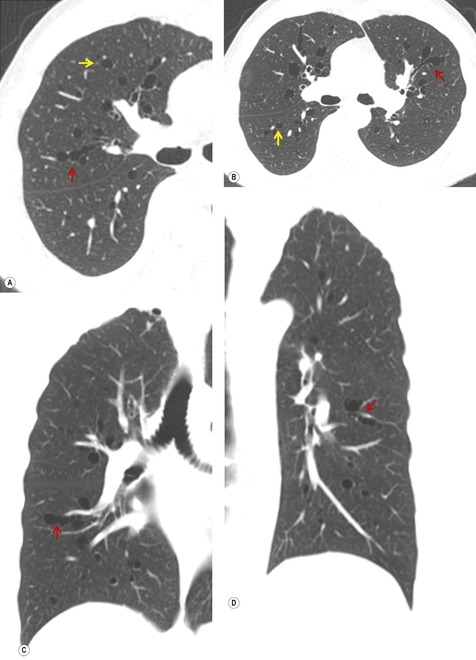
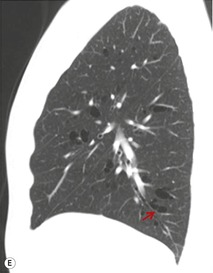
1. Central mucocele. Proximal bronchial atresia leads to accumulation of mucus within the bronchi distal to the atretic segment. The mucocele manifests on chest radiographs or computed tomography (CT) scans as a central masslike opacity. The opacity is typically round or oval in shape and may not have the branching configuration often associated with acquired mucoceles (Figs 16.5 and 16.6). There may, however, be blunt horn-like protrusions from the main mass.
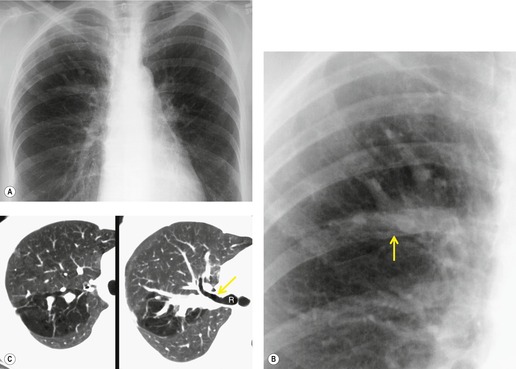
Almost 60% of patients with bronchial atresia are asymptomatic. In these patients, the mass is an incidental finding on a routine chest radiograph. Most are seen in young patients and there is a female predominance of 2:1. Symptomatic patients usually present with recurrent chest infections, dyspnea, wheezing, and chronic cough.
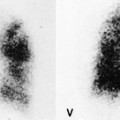
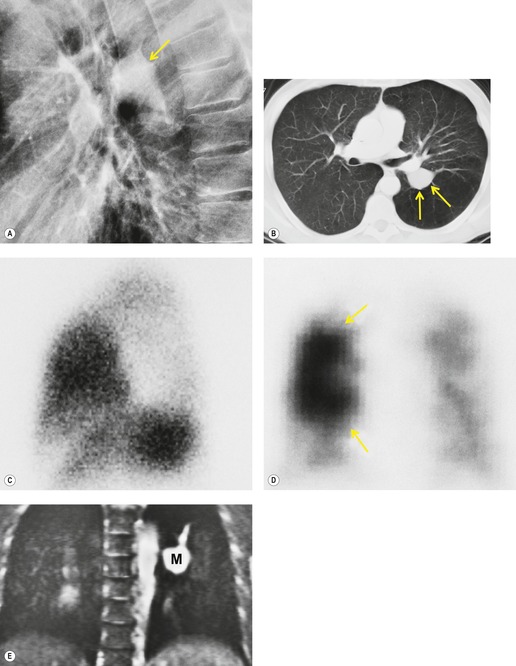
Meng et al. 41 reviewed 36 cases of bronchial atresia and found that 24 (66%) occurred in the left upper lobe and seven (19%) occurred in the right upper lobe. In the affected upper lobes, it is almost invariably the apical or apicoposterior segments that are involved. Tederlinic, 42 however, reported four cases of bronchial atresia in the lower lobes. CT is useful for demonstrating absence of the affected bronchus, the central mucus plug, and the surrounding decreased attenuation and hypoperfused lung.38. and 43. Radionuclide ventilation/perfusion scanning, now rarely performed for the diagnosis of bronchial atresia, can also demonstrate hypoperfusion and delayed air entry with air trapping in the affected portion of lung (Fig. 16.6). The central mucus plug is typically of high signal intensity on T2-weighted magnetic resonance (MR) images37. and 44. (Fig. 16.6). Bronchial atresia is of little clinical significance. Its importance is that the dilated, mucus-filled bronchus distal to the atresia should not be misdiagnosed as a neoplasm. 45
CONGENITAL LOBAR OVERINFLATION
Also known as congenital lobar emphysema, congenital lobar overinflation (CLO; Box 16.3) is characterized by progressive hyperexpansion of a lobe of lung. CLO is the preferred term because it best describes the underlying pathophysiology of this disorder – overinflation of normal alveoli, probably due to central obstruction. 46 Although the precise cause of obstruction has been difficult to determine in resected lung specimens, aplasia, hypoplasia, or dysplasia of bronchial support structures is postulated as the primary cause. The congenital nature of the condition has never been unequivocally established even though the circumstantial evidence is strong. There is a definite association with congenital heart disease. 47 A clinically identical but pathologically distinct condition, the polyalveolar lobe, has been described.48.49. and 50. In this condition the airways and the vascular supply to the involved lobe are normal. The lobe contains three to five times the normal number of alveoli for the patient’s age. The result is a relatively large lobe that may have all of the compressive effects seen with CLO. This giant lobe is not emphysematous. However, in practical clinical terms the condition is indistinguishable from true lobar overinflation.
Box 16.3
Cause
• Presumed to be bronchial obstruction
Presentation
• 90% of cases present before 6 months old
• Most present in the neonatal period
• Respiratory distress
Lobes involved
• Upper and middle lobes (less than 2% have lower lobe involvement)
• Left slightly more common than right
Imaging findings
• In utero (rare) – unilateral echogenic ‘mass’ (fluid in an obstructed lobe)
• Chest radiographs
– Before air enters the involved lobe – a homogeneously dense unilateral ‘mass’
– As air is entering and fluid is draining – focal streaky or bubbly mass lesion in one hemithorax. Rarely seen
– Most common manifestation: hyperexpansion and hyperlucency of a lobe with mediastinal shift and compression of adjacent lobes
Differential diagnosis
• Pneumothorax – localized especially anteriorly
• Unilateral or lobar pulmonary interstitial emphysema
• Reversible cause of obstructive overinflation, e.g. mucus plug
• Polyalveolar lobe
In most cases, congenital lobar overinflation manifests in the neonatal period with respiratory distress that may be life-threatening. In up to 25% of cases, however, presentation is delayed until after the first month of life. The classic and most common radiographic appearance is hyperexpansion of a single lobe of lung, usually an upper or middle lobe (Figs 16.7 and 16.8). 51 Remarkably, the lower lobes are involved in only about 2% of cases. Involvement of more than one lobe is equally rare. The expanded lobe may compress the remainder of the lung, accompanied by displacement of the heart, mediastinum, and diaphragm. The primary differential diagnoses include acquired lobar overinflation due to an endobronchial mass (e.g. foreign body) or extrinsic airway compression, localized pneumothorax, an expanding lung cyst, and, rarely, congenital cystic adenomatoid malformation. Careful study of the chest radiographs should reveal the presence of blood vessels within the hyperexpanded lobe in cases of CLO. Pulmonary interstitial emphysema, an acquired condition most commonly seen in neonates receiving positive-pressure ventilation, may affect a single lobe and closely resemble CLO radiographically. The conditions differ clinically, however, in that interstitial emphysema usually occurs in children who are already receiving mechanical ventilation because of previously known diffuse lung disease. Endobronchial foreign bodies, mucus plugs, or very rarely tumors can result in obstructive overinflation of a lobe that can appear radiographically identical to CLO. Bronchoscopy to exclude an obstructing lesion should therefore be performed prior to surgical resection of an overexpanded lobe.
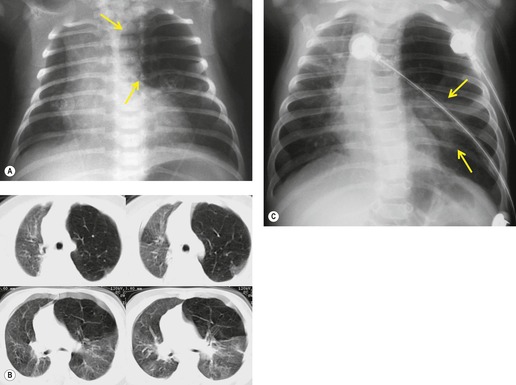 |
| Fig. 16.7 (Courtesy of JR Galvin, MD, Armed Forces Institute of Pathology, Washington, DC, USA.) |
Much less commonly, CLO can manifest as a hyperinflated, fluid-filled lobe. 52 On radiographic examination, the fluid-filled lobe is seen as an expansile mass of soft tissue density (Fig. 16.9). Compression of the remainder of the lung and displacement of the mediastinum may occur exactly as seen with the more common lucent form. In most of these cases, the entrapped fluid drains shortly after birth with resultant conversion to the more conventional appearance of CLO. On rare occasions, a transitional phase between the fluid-filled and the air-filled forms may be encountered. 53 In this transitory phase there are linear interstitial markings resembling septal lines in the hyperexpanded lobe. These opacities disappear, presumably as fluid drains or is absorbed.
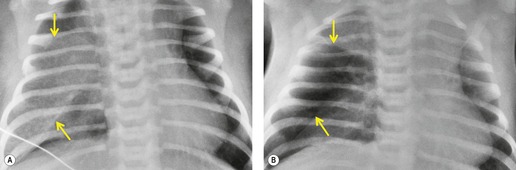
The diagnosis of congenital lobar overinflation is usually made on the basis of the clinical and chest radiographic findings. 51 CT may be performed to exclude alternative diagnoses such as primary pulmonary hypoplasia or scimitar syndrome).54.55.56. and 57. CT is useful for confirming the presence of lobar overinflation, for better delineating mass effect on adjacent structures, and for excluding endobronchial masses. 51 CT angiography can also be useful for differentiating CLO from various forms of pulmonary hypoplasia.
Resection of the affected lobe is the most common treatment for infants less than 2 months old and for patients of any age who have severe respiratory distress related to the overinflated lobe. 51 Patients older than 2 months who have mild to moderate symptoms and normal bronchoscopic examination are frequently managed conservatively51 with close radiographic follow-up. 58 Kennedy and co-workers54 followed up a group of 12 patients treated conservatively for up to 12 years. They found steady, gradual symptomatic improvement in all cases paralleled by a relative decrease in the lobar hyperexpansion on serial chest radiographs.
ABSENCE (AGENESIS OR APLASIA) OF LUNG OR LOBES OF LUNG (Table 16.1)
Unilateral absence of a lung or a lobe is a rare congenital abnormality that, in itself, causes surprisingly few clinical problems.59.60. and 61. Indeed, absence of a lung may be encountered de novo in an adult who has had no symptoms referable to the chest. However, the condition is frequently associated with other congenital abnormalities, particularly tracheoesophageal fistula and the VACTEL syndrome (vertebral, anal, cardiac, tracheoesophageal and limb).62.63. and 64. When the bronchus to the affected lung is absent, the condition is termed pulmonary agenesis; when there is a rudimentary bronchus, the condition is termed pulmonary aplasia. 65 The ipsilateral pulmonary artery develops but tends to be small or rudimentary. 66 Nearly 10% of patients with tracheobronchial malformations have unilateral pulmonary agenesis. Interestingly, agenesis of the right lung is associated with esophageal atresia67.68. and 69. whereas agenesis of the left lung is associated with isolated tracheoesophageal fistula without esophageal atresia. 60 Agenesis of the right lung is said to be twice as common as agenesis of the left lung. 70 Absence of one or more lobes of a lung is less common and, when present, is more common on the right. Absence of individual lobes is a form of hypogenetic lung syndrome, which may occur as an isolated entity simulating pulmonary hypoplasia or in association with anomalous pulmonary venous drainage as part of the venolobar syndrome (scimitar syndrome). 66
| VB, defect of the vascular bud; LB, defect of the lung bud. | ||
| Abnormality | Associated anomalies | Chest radiograph |
|---|---|---|
| Aplasia of a pulmonary artery (VB) | ||
| Slight lung volume reduction, small hilum and diminished vascularity, contralateral increased vascularity | ||
| Isolated | Right aortic arch | |
| Associated with hypogenetic lung syndrome | See below | See below |
| Associated with hypoplastic lung | See below | See below |
| Lobar agenesis/aplasia (LB) | ||
| Isolated absence of a lobe of lung | Usually none | Volume reduction of the affected lung |
| Mediastinal shift | ||
| Retrosternal density | ||
| Hypoplasia of lung (LB) | ||
| Primary | Absent pulmonary artery (VB) | Volume reduction of the affected lung |
| Cardiac anomalies | Mediastinal shift | |
| Retrosternal density | ||
| Secondary | See Box 16.4 | |
| Lung agenesis/aplasia (LB) | Frequent – | No visible aerated lung |
| Agenesis – complete absence of a lung and bronchus | Tracheoesophageal | Marked mediastinal shift and cardiac rotation |
| Aplasia – complete absence of a lung but rudimentary bronchus present | SkeletalGenitourinaryCardiacGastrointestinal | Herniation of the opposite lungElevated diaphragm |
| Scimitar syndrome (VB and LB) | Frequent – | Right lung only – |
| Horseshoe lung | Small volume lung | |
| Accessory diaphragm | Dextroposition of the heart | |
| Diaphragmatic defects | Stunted and deformed vascular pedicle | |
| Interrupted inferior vena cava | Scimitar vein(s) | |
| Sequestration | ||
| Cardiac | ||
| Systemic arterial supply to lung | ||
As might be expected, chest radiographs in patients with unilateral absence of a lung show loss of aeration and marked volume loss on the affected side (Figs 16.10 and 16.11). Volume loss is shown as elevation of the ipsilateral hemidiaphragm, shift of the mediastinum toward the abnormal side, and anterior herniation of the contralateral lung. There is usually a characteristic wedge-shaped opacity in the inferior and lateral aspect of the affected hemithorax (Fig. 16.11). This opacity is due to herniation of heart and mediastinum into the lower thorax and proliferation of fat to partially compensate for the absence of lung tissue. The obvious differential diagnoses are acquired total lung collapse or prior pneumonectomy. Further investigation with CT or bronchoscopy may be necessary to make the distinction (Fig. 16.11).
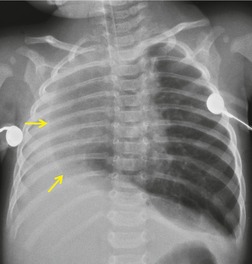
In cases of isolated absence of a lobe, there is compensatory hyperexpansion of the remaining lobe or lobes with consequent distortion of the bronchovascular structures. The compensatory expansion is never complete, and lung volume is reduced on the affected side. The chest radiographic findings in congenital absence of a lobe are indistinguishable from those of generalized pulmonary hypoplasia and are discussed below. CT, or now less commonly bronchography, can identify absence of a lobe by demonstrating agenesis or aplasia of the lobar bronchus with an otherwise normal airway. 65 In cases of pulmonary hypoplasia, on the other hand, the number of lobes is normal but the bronchial tree is stunted and deformed.
PULMONARY HYPOPLASIA (Table 16.1 and Box 16.4)
Pulmonary hypoplasia can be categorized as unilateral or bilateral and as primary (occurring de novo) or secondary (caused by other fetal developmental anomalies). 71
Box 16.4
Pressure effects on developing lung
• Pulmonary sequestration
• Cystic adenomatoid malformation
• Congenital diaphragmatic hernia
• Diaphragmatic eventration
• Chylothorax
• Hydrops fetalis
• Ascites or abdominal mass
Conditions associated with reduced or absent fetal respiration
• Central nervous system lesions
– Anencephaly
– Arnold–Chiari malformation
• Pena–Shokeir syndrome81
• Phrenic nerve agenesis
• Congenital myotonic dystrophy
Restrictive abnormalities of the thoracic cage
• Asphyxiating thoracic dystrophy
• Achondroplasia
• Thanatophoric dwarfism
• Ellis–van Creveld syndrome
• Osteogenesis imperfecta
Decreased pulmonary vascular perfusion
• Bilateral
– Ebstein anomaly
– Hypoplastic right heart syndrome
– Pulmonary stenosis
• Unilateral
– Pulmonary artery agenesis
– Scimitar syndrome
• Chromosomal abnormalities
– Trisomy 13, 18, and 21
Oligohydramnios
• Decreased urine output
– Renal agenesis
– Bladder outlet obstruction
• Amniotic fluid leak
Primary unilateral pulmonary hypoplasia
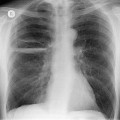
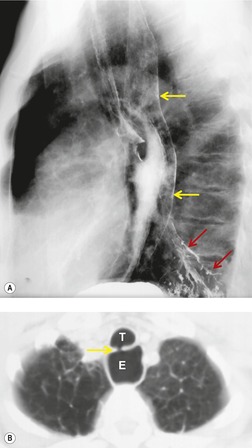
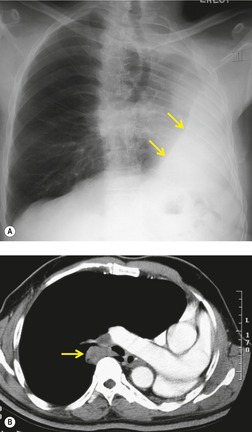
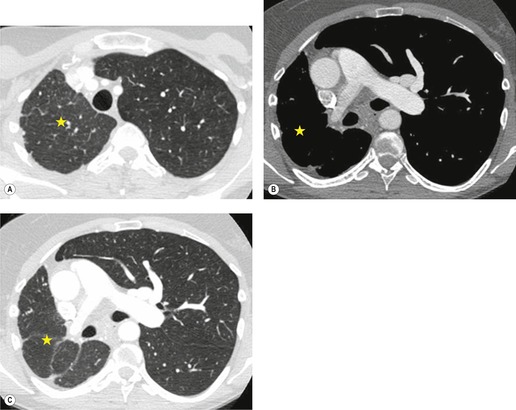
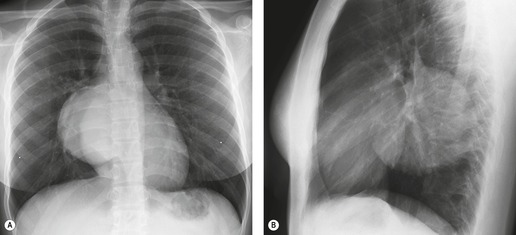
Primary unilateral pulmonary hypoplasia is usually associated with scimitar syndrome or other vascular anomalies (see below). 72 It may be encountered in either a child or an adult. Associated anomalies may be present in other body systems and affect outcome. Affected patients may be asymptomatic or have recurrent episodes of wheezing and pneumonia. The affected lung is small and the mediastinum is displaced into the ipsilateral hemithorax. The pulmonary vasculature is typically deformed and diminutive. The lateral chest film often shows a sharply marginated opacity behind and parallel to the sternum. The appearance is similar to that seen in cases of left upper lobe collapse or combined right upper and middle lobe collapse, but no wedge or fan of tissue extending to the hilum is seen (Figs 16.12 and 16.13). Previously, it was thought that the retrosternal opacity represented extrapleural areolar tissue filling the space that should have been occupied by lung. CT has shown that the density is produced by displacement of the heart and mediastinum into the ipsilateral thorax. 73 Cardiac contours may be indistinct on the frontal examination because of rotation of the heart and mediastinum.
Primary bilateral pulmonary hypoplasia
Primary bilateral pulmonary hypoplasia is quite rare and invariably manifests early in the neonatal period. Radiographs reveal small but otherwise aerated lungs with a bell-shaped thorax due to a marked difference in the size of the thorax and abdomen. 74 Pneumothoraces are frequently present.
Secondary pulmonary hypoplasia
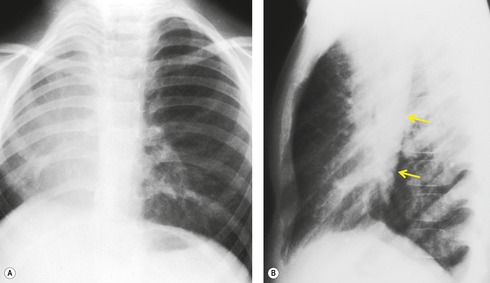
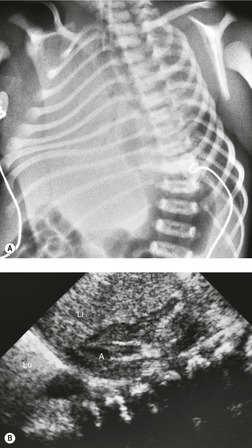
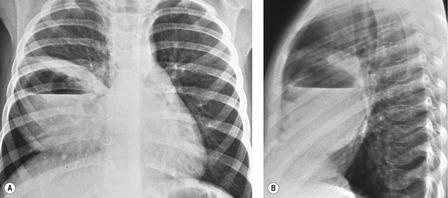
Overall, pulmonary hypoplasia caused by various fetal developmental abnormalities (secondary pulmonary hypoplasia, see Box 16.4) is more common than primary pulmonary hypoplasia. Pulmonary hypoplasia is found in approximately 10% of neonatal autopsies and in 50% of those with congenital anomalies. The mechanisms that cause pulmonary hypoplasia have not been firmly established. The fetal lung communicates freely with the amniotic sac and the fetus is known to exhibit breathing movements in utero. Thus, entry of amniotic fluid into the lungs probably occurs during fetal life. The fetal lung itself produces fluid as evidenced by accumulation of fluid beyond a bronchial obstruction. The presence of lung fluid and respiratory movement appears to play a vital role in normal lung development. Decreased or absent respiratory movement in utero caused by central nervous system anomalies is associated with pulmonary hypoplasia. 75 Decreased vascular perfusion of the fetal lung in cases of Ebstein anomaly, hypoplastic right heart syndrome, or pulmonary stenosis may impair fetal lung fluid production and be associated with hypoplasia. The most striking example of the influence of fluid on pulmonary development is found in Potter syndrome (oligohydramnios tetrad). 76 Oligohydramnios in these infants is the result of an abnormality in the development of the urinary tract, most commonly renal agenesis (Fig. 16.14). The full tetrad comprises: the underlying renal abnormality that causes oligohydramnios; abnormal facies resulting from pressure effects in utero; abnormal laxity of the skin; and pulmonary hypoplasia. 77 There is debate as to whether pulmonary hypoplasia results from a reduction in the amount of amniotic fluid actually within the developing lung or whether it is secondary to external pressure effects on the developing thorax. Clearly both factors could be operative.
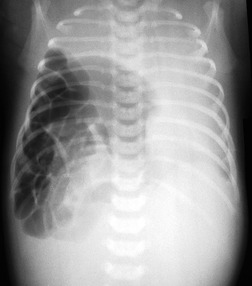
Pulmonary hypoplasia can also occur secondary to in utero conditions that stunt lung growth by virtue of mass effect. Pulmonary hypoplasia complicating congenital diaphragmatic hernia is a classic example (Fig. 16.15). The ipsilateral lung shows varying degrees of hypoplasia depending on the size of the hernia and the period during which pressure effects on the lung were operative. The contralateral lung may also have a variable degree of hypoplasia depending on the amount of mediastinal shift. The fundamental problem in such cases is lack of space for growth and development of the lung. The extent of pulmonary hypoplasia is a critical determinant of survival following surgical repair of diaphragmatic hernia or removal of intrathoracic mass lesions. 78 Chest radiographs can be helpful for assessing the degree of hypoplasia and for predicting prognosis in affected infants.78. and 79. The radiographic manifestations of secondary pulmonary hypoplasia are the same as those of primary hypoplasia, with the exception that the cause (e.g. congenital diaphragmatic hernia, cystic adenomatoid malformation, etc.) may also be identifiable. Rib crowding and deformity may give an indication of intrauterine pressure effects as in cases of Potter syndrome. Respiratory distress is common and the patients are often intubated. Pneumothoraces often complicate the clinical course. Ultrasonography has shown promising results in the antenatal diagnosis of pulmonary hypoplasia. 80
SCIMITAR SYNDROME (see Table 16.1 and Box 16.5)
Scimitar syndrome is sometimes termed the ‘hypogenetic lung syndrome’ or the ‘venolobar syndrome’ to emphasize that this anomaly is not simply a variant of pulmonary venous return but is a rather more extensive malformation. Felson65 coined the term venolobar syndrome and Woodring et al. 70 extended the use of this term to encompass a range of anomalies including pulmonary hypoplasia and sequestration. However, pulmonary hypoplasia and lobar aplasia can occur in the absence of anomalously draining veins. Therefore, the term ‘scimitar syndrome’ may be the most unambiguous designation of this form of pulmonary dysmorphism, recognizing that cases that deviate from the classic description of the condition are rare.70.82.83. and 84.
Box 16.5
Also termed:
• Venolobar syndrome
• Hypogenetic lung syndrome
Associations
• Congenital heart disease in 25%
• Usually atrial septal defects
Symptoms
• Usually asymptomatic
• Dyspnea if large left-to-right shunt
Imaging findings
• Small right lung (pulmonary hypoplasia)
• Diminutive right hilum
• Hyparterial right bronchus
• Dextroposition of heart
• Characteristic ‘scimitar’ veins draining below diaphragm
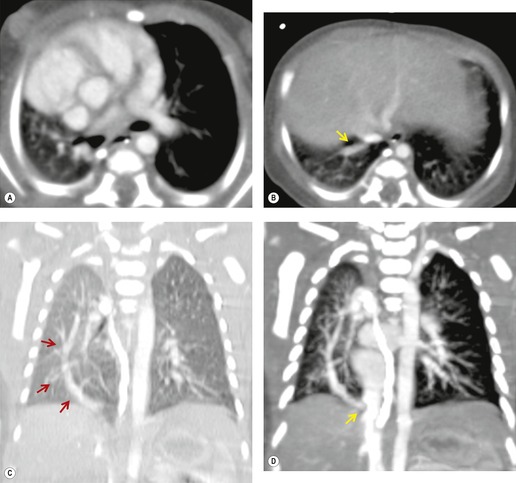
Scimitar syndrome is essentially an anomaly of the right lung. Only rare cases have been described on the left. 85 The affected lung is hypoplastic, with underdevelopment of both the central airways and vasculature.86. and 87. One or more lobes may be absent. The airway on the involved side is stunted and the arterial supply to the right lung may derive in much greater proportion than normal from systemic arteries. The pulmonary artery is correspondingly small and may lie beneath the right bronchus. The syndrome derives its name from the anomalous pulmonary vein that descends vertically in the lung before curving medially to enter the inferior vena cava above or below the diaphragm. The vein broadens as it curves downward and medially, resulting in a configuration resembling a Turkish sword – the scimitar (Figs 16.16 and 16.17). On occasion, two or more anomalous veins are present. The anomalous vein may drain all or part of the involved lung and empties into the inferior vena cava, the right atrium, coronary sinus, or, rarely, the hepatic veins.
Approximately 25% of patients have associated congenital heart disease, most commonly septal defects. 88 Such cases are more likely to present in infancy or childhood and the outcome depends on the type and severity of the cardiac abnormality. 89 The scimitar syndrome, when isolated, is compatible with a normal life because the consequent left-to-right shunt is typically small. 90 Occasionally, the shunt is large enough to warrant surgical repair. 91 Patients occasionally have symptoms from associated bronchiectasis and tracheal diverticula. Other associated anomalies include eventration or Bochdalek hernia involving the right hemidiaphragm, accessory right hemidiaphragm, and horseshoe lung (see below).
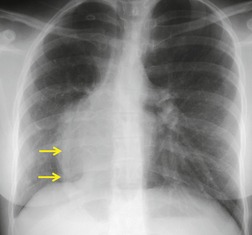
The anomalously draining vein is usually readily visible on chest radiographs in both the frontal and the lateral projections (Figs 16.16 and 16.17). Associated features include reduced lung volume, shift of trachea and mediastinum to the right, consequent dextroposition of the heart, a diminutive ipsilateral pulmonary artery, and an abnormal bronchovascular pattern in the right hilum. When all of these findings are present, the diagnosis of scimitar syndrome can be made with confidence (Figs 16.16 and 16.17). Partial anomalous pulmonary venous, returning to the inferior vena cava, right atrium, or coronary sinus, may occur without pulmonary hypoplasia, particularly in association with intracardiac lesions such as an atrial septal defect or complex cyanotic lesions. The key distinguishing feature in these cases is the absence of radiographic evidence of pulmonary hypoplasia.
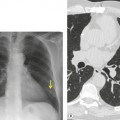
Contrast-enhanced CT provides a noninvasive method for confirming the diagnosis of the scimitar syndrome and identifying the termination of the anomalous veins (Fig. 16.17).88.92.93.94.95. and 96. It has the additional advantage that the tracheobronchial anomalies are better delineated and tracheal diverticula and bronchiectasis are readily detected. Magnetic resonance imaging (MRI) has been used to both diagnose scimitar syndrome97.98.99.100. and 101. and to estimate the degree of left-to-right shunting by means of velocity-encoded cine MRI. 102 CT may also demonstrate the presence of horseshoe lung, a rare associated abnormality. 90 Horseshoe lung is an uncommon malformation where right and left lungs are fused inferiorly by an isthmus of lung tissue crossing the posterior mediastinum.103.104.105.106.107. and 108. Horseshoe lung occurring in the absence of scimitar syndrome is often associated with other lethal malformations. Scimitar syndrome can sometimes be confused radiographically with unilateral agenesis of the pulmonary artery (see below) when systemic collateral vessels simulate scimitar veins. 82 Furthermore, an abnormal ‘meandering’ vein draining a hypoplastic right lung to the left atrium can resemble a scimitar vein. 70
UNILATERAL ABSENCE OF THE PULMONARY ARTERY
Unilateral absence of the pulmonary artery (UAPA) is a rare anomaly characterized by short segment atresia of the proximal left or right pulmonary artery.109.110.111.112.113.114. and 115. More distal segments of the arteries in the hila are usually present. The condition is associated with other congenital anomalies, particularly cardiac defects such as tetralogy of Fallot, septal defects, and pulmonary stenosis. 111 Isolated UAPA also occurs. The atretic segment usually, but not always, occurs on the side opposite to the aortic arch.
The majority of patients with isolated UAPA are symptomatic at presentation. In a review of 108 patients with isolated UAPA, Ten Harkel et al. 111 found that 87% presented with symptoms that included frequent pulmonary infection (37%), dyspnea or limited exercise tolerance (40%), or hemoptysis (20%). Pulmonary hypertension was present in 44% of the patients. Only 13% were asymptomatic. 111 Hemoptysis is due to systemic collateralization of the lungs.111.114. and 115. The etiology of pulmonary hypertension in these individuals is unknown.
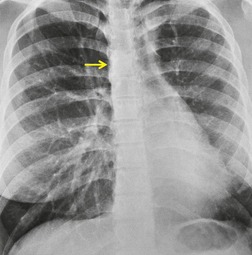
Chest radiographs show a reduction in lung volume in the affected hemothorax. The pulmonary hilum is typically diminutive or absent and peripheral perfusion is typically reduced (Figs 16.18 and 16.19). On occasion, increased reticular opacities are noted in the affected lung due to systemic-to-pulmonary collaterals. The vasculature in the normal lung may appear correspondingly plethoric because the entire cardiac output is shunted through that lung. CT scanning or MRI clearly shows the absence of a pulmonary artery and may also demonstrate evidence of systemic-to-pulmonary arterial collateral vessels.116. and 117. CT or MRI should obviate the need for angiography (Fig. 16.19). Acquired conditions such as Swyer–James syndrome or fibrosing mediastinitis may, however, closely mimic the findings of UAPA in adults.118. and 119. Swyer–James syndrome (see p. 750) should have demonstrably abnormal ventilation of the affected lung. Lung ventilation is normal in cases of UAPA. Fibrosing mediastinitis may sometimes be difficult to distinguish from UAPA on imaging. The presence of calcified lymph nodes in the vicinity of the occluded vessel or an associated mediastinal mass suggests fibrosing mediastinitis. 120
PULMONARY ARTERIOVENOUS MALFORMATIONS
Pulmonary arteriovenous malformations (PAVMs; Box 16.6) result from abnormal communication between the pulmonary arteries and veins. PAVMs are twice as common in females as in males and the vast majority of lesions are congenital in origin. According to Gossage et al., 121 between 53% and 70% of PAVMs occur in the lower lobes of lung. In a pathologic review of 350 autopsies, Bosher et al. 122 found that 75% of affected individuals had unilateral PAVMs, 36% had multiple lesions, and that 50% of those with multiple lesions had bilateral PAVMs. Nearly 70% of cases are associated with hereditary hemorrhagic telangiectasia (HHT or Rendu–Osler–Weber disease), and, especially in this disease, the lesions are likely to be multiple. 121 The Rendu–Osler–Weber syndrome is also known as hereditary hemorrhagic telangiectasia. It is an autosomal dominant disorder typically identified by the triad: telangiectasia, recurrent epistaxis, and family history for the disorder. The genetic defect causing the disease involves the ENG or ALK-1 genes. The major cause of morbidity and mortality is the presence of multiorgan arteriovenous malformations. The potentially resulting episodes of hemorrhage may be life-threatening. The disease has a wide spectrum of presentations, ranging from asymptomatic stages to severe multiorgan involvement. The treatment consists in the surgical management of bleeding. The prognosis of the disease depends on the severity of symptoms. Patients are at risk for both mucosal and visceral hemorrhage, for cardiac failure, cerebral abscesses, ischemic stroke, and migraines. The worldwide prevalence is 1 case per 5000–10 000. Chest radiography is recommended as a screening test for pulmonary arteriovenous malformations in patients with the disease. All patients with low-grade evidence of pulmonary abnormalities require CT as confirmatory study. CT may be complemented by angiography, notably in cases of planned angiographic intervention.123.124.125.126. and 127. PAVMs in hereditary hemorrhagic telangiectasia may also be associated with other congenital abnormalities, notably cardiac malformations.128. and 129.
Box 16.6
Single or multiple lesions. Majority associated with HHT syndrome – an autosomal dominant disorder with incomplete penetrance.
Clinical presentation
• Often asymptomatic
• May present with other manifestations of HHT, e.g. epistaxis, gastrointestinal bleeding
• Hypoxia and orthodeoxia with dyspnea and chronic fatigue
• Embolic phenomena – brain abscesses and cerebral ischemia
• Clubbing and polycythemia
• Rarely hemoptysis
• Rarely cyanosis and a chest bruit in infants
Treatment
• Feeding vessel <3 mm – not generally treated
• Feeding vessel >3 mm – occlusion of feeding vessels with detachable balloons or coils
• Surgery reserved for very large lesions
Infants and children with PAVMs may be asymptomatic. However, if the lesions are large enough to cause a marked right-to-left shunt, the patient may present with cyanosis or heart failure (Fig. 16.20). In such cases, a distinct bruit may be heard over the affected hemithorax. Many adult patients with PAVMs are asymptomatic when the lesions are detected on routine chest radiographs. There is evidence, however, that the likelihood of developing symptoms related to PAVMs depends upon the size and number of lesions. 121 Thus, patients with a solitary PAVM larger than 2 cm in diameter or patients with multiple PAVMs are more likely to have symptoms than those with small, solitary lesions. When present, signs and symptoms include dyspnea, cyanosis, clubbing, hemoptysis and chest pain. On occasion, the patient may present with systemic abscesses or infarction, notably of the brain, because right-to-left shunting through the PAVM bypasses the lung (paradoxical embolism, Fig. 16.21).130.131. and 132. One-third of patients in one large series had CT evidence of previous cerebral infarction. 133 On physical examination, patients with HHT and PAVM may have superficial, often perioral, telangiectasias. A bruit is heard over the lesion in up to 46% of patients. 121 Even in asymptomatic adults, right-to-left shunting though the lesion often produces some degree of hypoxia that is often accentuated in the erect position. This finding is known as orthodeoxia, is characterized by detecting a decrease in arterial oxygen saturation when the patient moves from the supine to erect position, and is a typical feature of PAVM. The physiologic explanation for this phenomenon is that the lesions have a predilection for the lower lungs and that shunting is therefore increased in the upright position. 121
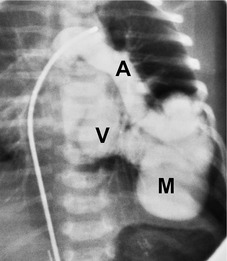
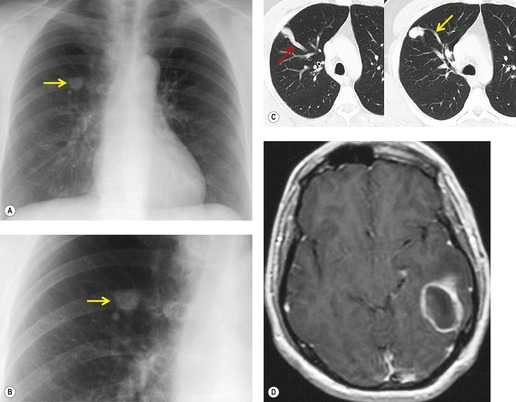
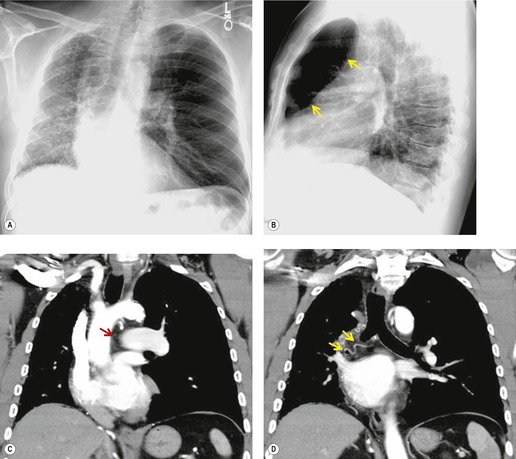
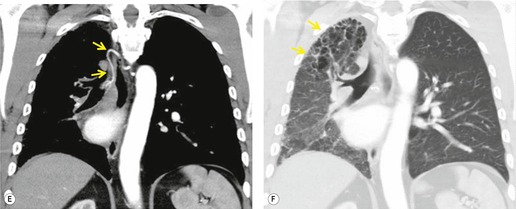
On chest radiographs, arteriovenous malformations manifest as well-circumscribed lung nodules, usually with a lobular contour (Fig. 16.21). The lesions typically range in size from 1 cm to 5 cm in diameter. 121 If the lesions are peripheral in location, the feeding arteries and draining veins can usually be seen on the chest radiographs (Figs 16.21 and 16.22) – a diagnostic feature that can be confirmed by CT or MRI. Central lesions are often more difficult to discern among the hilar vessels on chest radiographs (Fig. 16.23), and, even if the lesion itself is detected, the shorter feeding vessels may be difficult or impossible to identify. Multiple lesions can be confused with metastases if the feeding vessels are overlooked. 134 In patients with suspected PAVM, contrast-enhanced echocardiography121 can be used to confirm the presence of an intrapulmonary right-to-left shunt. Perfusion scintigraphy can also be used to diagnose and, more importantly, quantify the right-to-left shunt.135.136. and 137. Perfusion scintigraphy cannot, however, distinguish intracardiac from intrapulmonary shunting and thus is used infrequently for diagnosis of PAVM.
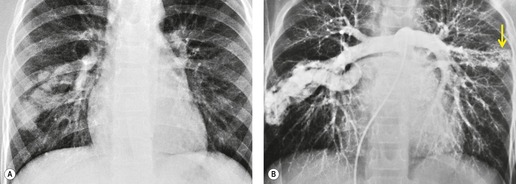
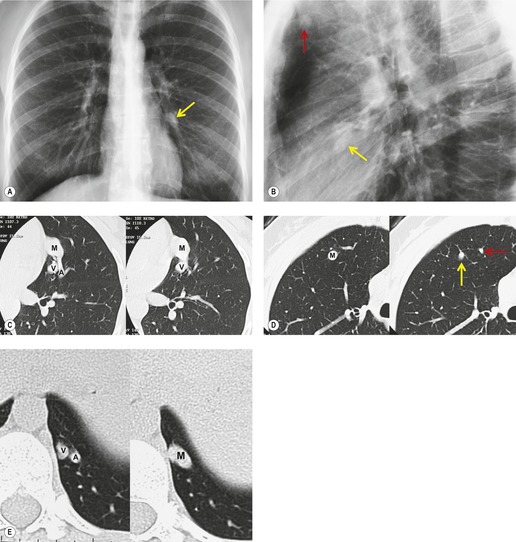
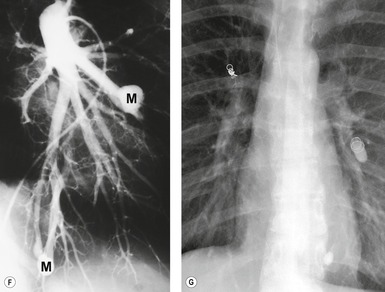
Although pulmonary angiography has been considered the ‘gold standard’ for diagnosis of PAVMs (Figs 16.22 and 16.23), 121 CT has recently assumed a larger role in this regard.138.139.140.141.142. and 143. Contrast-enhanced CT is quite useful for confirming the diagnosis by demonstrating the feeding vessels and intense contrast enhancement within these lesions. CT is also useful for identifying additional lesions in the lungs (Fig. 16.23). Interestingly, Remy and co-workers140 found that CT was more sensitive than pulmonary angiography for detecting PAVMs. In their series, CT detected 98% of PAVMs as opposed to 60% detected by pulmonary angiography. Recently, contrast echocardiography has shown predictive of the presence and severity of PAVMs seen on CT. 144
Increasingly, PAVMs are treated by embolotherapy with coils,145.146.147. and 148. detachable balloons, or vascular plugs125.133.149. and 150. (Fig. 16.23). The purpose of such intervention is to reduce the risk of systemic embolization and to improve arterial oxygenation by decreasing right-to-left shunting. 151 Treatment of simple PAVMs (single feeding vessel, 80% of lesions) 152 is considered more straightforward than treatment of complex (multiple feeding vessels, 20%) lesions. 121 Furthermore, embolotherapy is usually restricted to malformations with feeding vessels 3 mm or greater in diameter. 133 Thus, information regarding size and number of feeding vessels is considered critical for planning embolotherapy of PAVMs. In past years, pulmonary angiography was performed for this purpose (Fig. 16.23). However, studies have shown that thin-section contrast-enhanced volumetric CT with 2-D and 3-D reconstruction techniques can be quite useful for delineating the often complex angioarchitecture of even small PAVMs (Fig. 16.24).142. and 143. Thus, angiography is now performed primarily for the treatment, not diagnosis, of PAVMs.
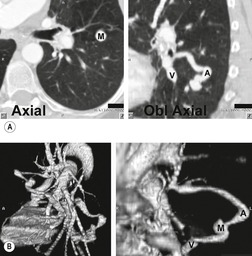
PULMONARY VARIX
A pulmonary varix is the enlargement of a pulmonary vein near its insertion into the left atrium. Most varices are congenital, with a minority of cases associated with pulmonary venous hypertension or hepatic cirrhosis. As a rare and often asymptomatic disorder, a pulmonary varix is often an incidental finding. In rare cases, patients may present with hemoptysis. The anomaly may rarely serve as a potential thrombogenic source and cause of atrial fibrillation.157. and 158.
AZYGOS ANEURYSM
Dilatation of the azygos vein may be caused by fluid overload and also, very rarely, by congenital azygos aneurysms. These aneurysms may represent the development of a remnant of an embryological vein that empties into the transverse part of the azygos vein. Aneurysms of the azygos vein are often asymptomatic, and are usually incidentally detected on a chest radiograph. If the aneurysm is very large, it may compress the right main bronchus or the superior vena cava. These aneurysms are detected with transesophageal echocardiography, CT, or MRI. The enhancement pattern of the lesion after administration of contrast material will depend on the presence and the degree of thrombosis. 159
CONGENITAL DISORDERS OF LYMPHATIC DEVELOPMENT
Faul et al. 160 have proposed the following classification for congenital disorders of lymphatic development (Box 16.7): thoracic lymphangioma, lymphangiectasis, lymphangiomatosis, and lymphatic dysplasia syndrome.
Box 16.7
Lymphangioma
• Focal neck mass
• 10% mediastinal
• Multicystic masses on CT and MRI
Lymphangiectasis
• Rare and frequently fatal
• Dilatation of otherwise normal lymphatics
• Associated with Turner, Noonan, Down syndromes
• Usually diffuse, but occasionally focal in lung
• Diffuse interlobular septal thickening, pleural fluid on CT
Lymphangiomatosis
• Diffuse proliferation of infiltrative lymphangiomas
• Rare and frequently fatal
• Chylothorax and cystic bone lesions are suggestive
• Diffuse interlobular septal thickening, pleural fluid on CT
Lymphatic dysplasia syndromes
• Primary lymphedema
• Congenital chylothorax
• Yellow nail syndrome
• Bronchiectasis (100%)
• Yellow nails (89%)
• Lymphedema (80%)
• Pleural (sometimes pericardial) effusions (36%)
• Lung cysts (rare)
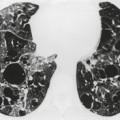
Lymphangiomas, or cystic hygromas, are focal masslike proliferations of lymphatic tissue160 that are classified histopathologically as capillary, cavernous, and cystic. The cystic spaces are typically filled with proteinaceous fluid. They usually present in early infancy, but adult cases are reported. 161 Most occur in the neck and axilla (Fig. 16.25), but 10% extend into the mediastinum and 1% occur only in the mediastinum. 162 On chest radiographs, lymphangiomas manifest as unilateral soft tissue masses in the neck that may demonstrate extension into the mediastinum. Most mediastinal lymphangiomas in adults are found in the anterior or superior mediastinum (Fig. 16.26). 163 Lymphangiomas manifest on ultrasound as uni- or multilocular cystic masses with walls of variable thickness. 164 They manifest on CT as smoothly marginated multicystic masses (Fig. 16.26);163. and 165. the cyst walls variably enhance following administration of intravenous contrast material. 164 In one study, the CT attenuation of the cyst fluid ranged from −4 HU to 34 HU, likely due to variability in lipid content and intracystic hemorrhage. 164 Unusual CT features include calcification, spiculated margins, and homogeneous soft tissue attenuation. 163 On T1-weighted MR images, the cysts are usually isointense to skeletal muscle. On T2-weighted MR images, they are typically hyperintense to fat (Fig. 16.25). 164 MR imaging more clearly delineates the extent of the lesion than ultrasound or CT.163.164.165. and 166.
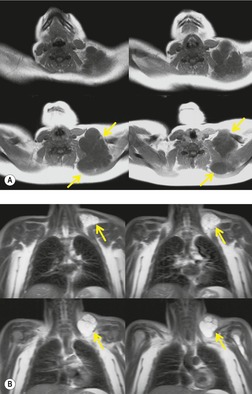 |
| Fig. 16.25 (Courtesy of J R Galvin, Armed Forces Institute of Pathology, Washington, DC, USA.) |
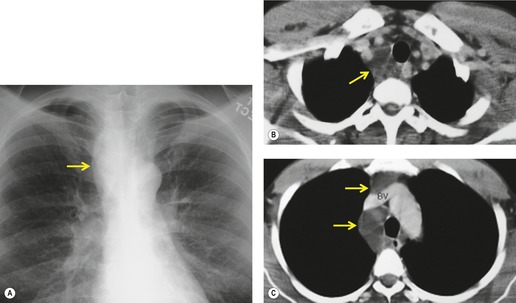 |
| Fig. 16.26 (Courtesy of E F Patz Jr, MD, Durham, NC, USA.) |
Generalized dilatation of otherwise histopathologically normal lymphatics is known as lymphangiectasis.160. and 167. The condition may occur primarily, or be secondary to severe pulmonary venous obstruction in cases of total anomalous pulmonary venous return or hypoplastic left heart syndrome. 168 Primary generalized lymphangiectasis is an extremely rare, lethal abnormality. At least half of affected individuals are stillborn and the remainder die shortly after birth. Lymphangiectasis can be confined to the lung169. and 170. and, in such cases, symptoms may be delayed in onset or absent. 171 Pulmonary lymphangiectasis is associated with several genetic diseases including Noonan, Turner, Ehlers–Danlos, and Down syndrome. 160 Chest radiographs in patients with pulmonary lymphangiectasis show diffuse reticulonodular opacities with thickened interlobular septa; the linear opacities may diminish over time if the patient survives (Fig. 16.27).171. and 172. Thin-section CT shows thickened interlobular septa and ground-glass opacities (Fig. 16.28); pleural effusions are also common. 173 Echocardiography is useful for excluding a cardiac etiology.
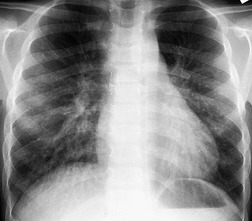
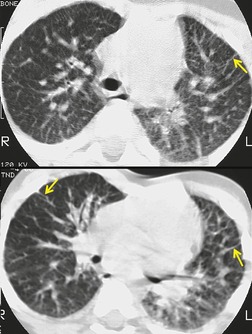
The presence of multiple, often infiltrating, lymphangiomas is termed lymphangiomatosis (Fig. 16.29). Lymphangiomatosis is often confused with lymphangiectasia, but the conditions are distinct histopathologically. 174 The condition is rare and often fatal. 160 It most often affects lungs and bones, but virtually any organ system can be involved.175.176.177. and 178. Cystic bone lesions and chylothorax suggest the diagnosis and portend a poor outcome.160. and 179. In a review of five patients, Wunderbaldinger et al. 179 found that the most common features of generalized lymphangiomatosis were widespread cystic lesions in visceral organs, lytic bone lesions, interstitial lung disease, and diffuse mesenteric thickening on CT or MRI. In a review of eight patients with pulmonary lymphangiomatosis, Swensen et al. 180 found that the disease most commonly manifested on thin-section CT scans with smooth thickening of interlobular septa, thickening of bronchovascular bundles, and scattered ground-glass opacities. Diffuse increase in mediastinal fat attenuation was a suggestive finding, seen in all eight patients. Seven of eight also had either bilateral pleural effusions or pleural thickening. The CT appearance of pulmonary lymphangiomatosis is virtually identical to that of pulmonary lymphangiectasia. 173
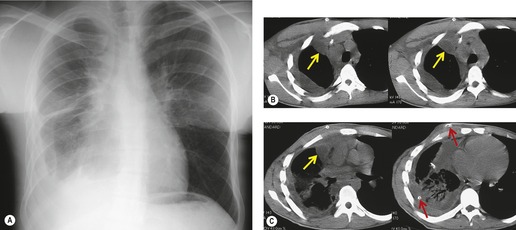 |
| Fig. 16.29 (Courtesy of E M Marom, MD, Houston, TX, USA.) |
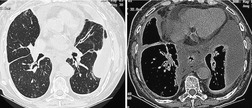
The term lymphatic dysplasia syndrome has been proposed to encompass the primary lymphedema syndromes, congenital chylothorax and the yellow nail syndrome. 160 This is a diverse group of disorders with variable prognosis. The yellow nail syndrome typically presents in adulthood; the median age at presentation is between 40 and 50 years.160.181. and 182. It is characterized by the triad of yellow nails (89%), lymphedema (80%), and pleural effusions (36%) (Fig. 16.30).183. and 184. The complete triad is seen in just 20% of patients, however.183. and 184. Thus, the presence of two of the three major findings is considered sufficient for diagnosis.184.185.186. and 187. The respiratory tract is affected in 63% of cases; manifestations include cough, shortness of breath, sinusitis and bronchiectasis. Wiggins et al. 188 evaluated four patients with thin-section CT and found bronchiectasis and bronchial wall thickening in all. Recurrent pericardial effusions and chylous ascites are less common manifestations.189. and 190. Chemical pleurodesis has been used to effectively treat symptomatic pleural effusions in affected patients.191. and 192. Cystic lower lobe lesions, not thought to represent bronchiectasis, have also been reported on CT. 193
BRONCHOGENIC CYST
Bronchogenic cysts (Box 16.8) are uncommon, usually single, lesions that result from abnormal budding of the ventral foregut between the 26th and 40th days of gestation. 194 This abnormal bud subsequently differentiates into a fluid-filled, blind-ending pouch. Bronchogenic cysts typically have a fibrous capsule, contain cartilage, are lined with respiratory epithelium (Fig. 16.31), and contain mucous material that may be remarkably viscid. Most cysts are found in the mediastinum or hila – near the tracheal carina (Fig. 16.32). In the four largest series reported, between 65% and 86% of cysts arose in the mediastinum.194.195.196. and 197. Less commonly, cysts may occur within the lung parenchyma (Fig. 16.33), pleura, or diaphragm. 194 Parenchymal cysts are usually perihilar in location (66%). In rare instances, bronchogenic cysts develop in ectopic locations such as the cervical, infradiaphragmatic, pericardial, and paravertebral regions. 198 Bronchogenic cysts are sometimes found in association with other congenital pulmonary malformations such as sequestration and lobar overinflation.194.199. and 200. Some pulmonary bronchogenic cysts have a systemic arterial supply and therefore may represent a form of pulmonary sequestration.201. and 202. Surgeons must consider the possibility of systemic arterial supply in cases requiring resection. 203
Box 16.8
Pathology
• Single cyst lined by respiratory epithelium
• Contains mucoid material
• Associated anomalies – rare
• Location – 85% hilar or mediastinal
Clinical features
• May be asymptomatic
• Otherwise chest pain, cough, dyspnea, and fever if cyst is infected
• Rarely compressive effects, e.g. atelectasis, postobstructive pneumonia
• Treatment – usually surgical excision
Chest radiographs
• Well-circumscribed hilar or middle mediastinal mass
• Air–fluid level if cyst is communicating or infected
• Less commonly, a well-circumscribed lung mass – solid or containing an air–fluid level.
Chest CT
• Well-circumscribed mass usually adjacent to trachea or major bronchi
• Molded to adjacent structures without remarkable compressive effects
• Variable CT density of cyst contents −10 HU to +120 HU or even higher
• About half of water, half of soft tissue attenuation
• Cyst wall thin and smooth
• Cyst may contain milk of calcium or wall calcification
• Gas content indicates communication or infection
MRI
• Well-circumscribed mass
• Variable signal intensity, depending on cyst content
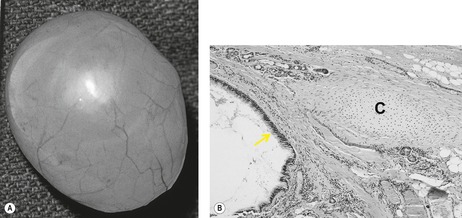 |
| Fig. 16.31 (With permission from McAdams HP, Kirejczyk WM, Rosado-de-Christenson ML, et al. Bronchogenic cyst: imaging features with clinical and histopathologic correlation. Radiology 2000;217:441–446.) |
Most patients with a bronchogenic cyst present in the first few decades of life.194. and 204. Presentation beyond 50 years of age is distinctly unusual. 205 The clinical features of bronchogenic cyst are variable. Infants may present with respiratory distress due to compression of the central airways.204. and 206. In such cases, the chest radiograph may show hyperexpansion of an entire lung as a result of a ball valve mechanism or collapse of a lobe or entire lung. 207 A mediastinal mass may be visible but may be masked by the thymus. Feeding difficulties caused by pressure effects on the esophagus or edema caused by impaired venous return are less common manifestations.208. and 209. Some older children and adults with bronchogenic cyst are asymptomatic at presentation; such lesions are detected on routine chest radiographs. 210 However, most older children and adults are symptomatic at presentation.194.196. and 204. St-Georges et al. 196 found that 70% of 86 patients had symptoms of chest pain (55%), cough (50%), dyspnea (40%), fever (30%), and purulent sputum (20%). Ribet et al. 204 found that 80% of adult patients with bronchogenic cyst had a similar pattern of symptoms. Occasionally, patients complain of dysphagia due to esophageal compression, and there are a few reports of pulmonary artery and vein obstruction by the cyst.211.212.213. and 214. Rarely, a bronchogenic cyst may undergo a rapid increase in size due to hemorrhage, infection, or distension with air. In such cases, the compressive effects of the enlarging cyst may cause a surgical emergency.206. and 207. There is a report of a peripheral bronchogenic cyst presenting with a spontaneous pneumothorax. 215
On chest radiographs, bronchogenic cysts manifest as well-defined, solitary mediastinal or hilar masses (Fig. 16.32); approximately 10% have a lobular contour. 195 They are usually found in close proximity to the major airways, and therefore one of the surfaces of the cyst usually contacts the trachea or central bronchi. Most cysts occur in the middle mediastinum immediately adjacent to the lower trachea or proximal main bronchi. Anterior and posterior mediastinal cysts are unusual.216. and 217. As the cysts enlarge, they displace adjacent lung and esophagus, but the central airways, except in very young children, are usually displaced little, if at all. Calcification, either along the rim or ‘milk of calcium’ within the cyst, is unusual.195.218. and 219. The cysts are usually stable in size except when complicated by infection or hemorrhage. 220 When infected, the cyst may become air-filled195. and 218. (Fig. 16.33). If the cyst contains air or an air–fluid level, the smooth thin wall and the central location of the cavity should indicate its nature and permit distinction from mediastinal abscess. Barium swallow examination is not usually performed for evaluation of mediastinal masses. If performed, perhaps for symptoms of dysphagia, barium swallow shows smooth extrinsic displacement of the esophagus by the cyst in at least half of cases. Typically, the cyst is seen between the airways and the esophagus on the lateral projection.
CT is now the standard technique for diagnosing bronchogenic cysts (Fig. 16.34, Fig. 16.35, Fig. 16.36 and Fig. 16.37). CT usually reveals a thin-walled cystic mass in the middle mediastinum, molded to the adjacent bronchovascular structures. 194 The molding of the cyst may cause one margin of the cyst to have a pointed configuration. 203 The cyst wall is smooth on both its inner and its outer margins and, in uncomplicated masses, is only a few millimeters thick. Most bronchogenic cysts are of water attenuation (−10 HU to +10 HU) on CT (Figs 16.33 and 16.34). 194 Although the majority are unilocular, multilocular bronchogenic cysts are rarely reported. 219 The CT diagnosis of a bronchogenic cyst can be accepted with confidence if the following criteria are met: a well-defined mass with a smooth or lobular outline, a uniformly thin wall, and contents of homogeneous CT attenuation within a range of −10 HU to +10 HU. However, a substantial minority of bronchogenic cysts are of soft tissue attenuation on CT (Fig. 16.36); 194 cysts with CT numbers as high as 120 HU have been reported. This finding on CT presumably reflects the fact that many cysts contain proteinaceous debris or hemorrhage.221. and 222. Rarely, a bronchogenic cyst contains milk of calcium, resulting in extremely high CT density223 (Fig. 16.37).
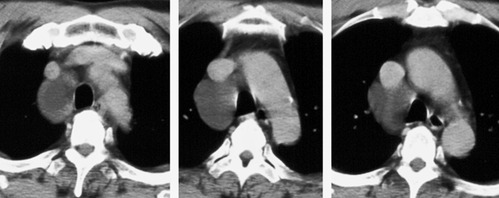
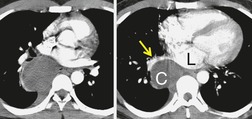 |
Fig. 16.35
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|