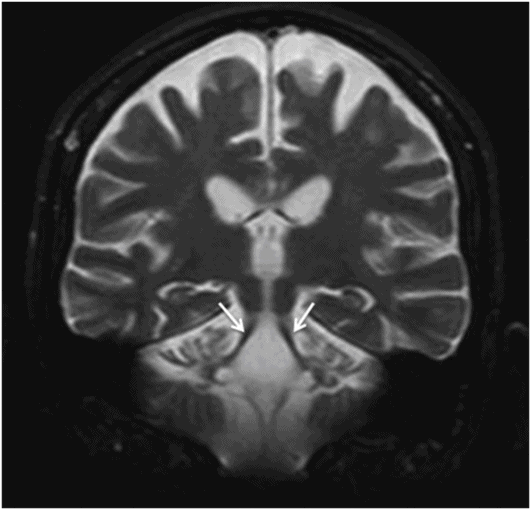
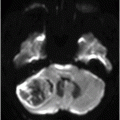
Coronal T2WI of the brain through superior cerebellar peduncles.
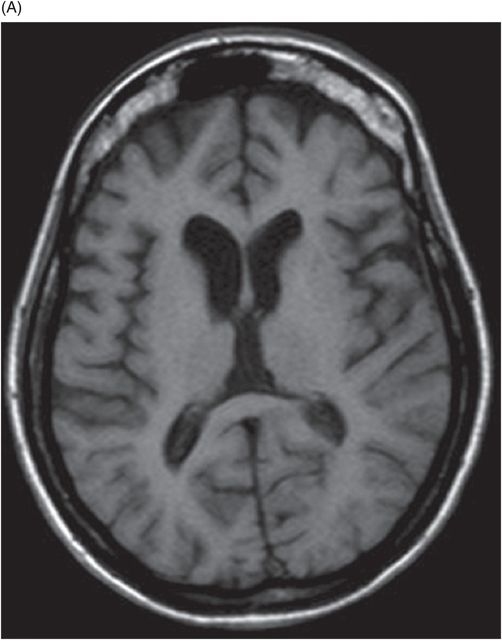
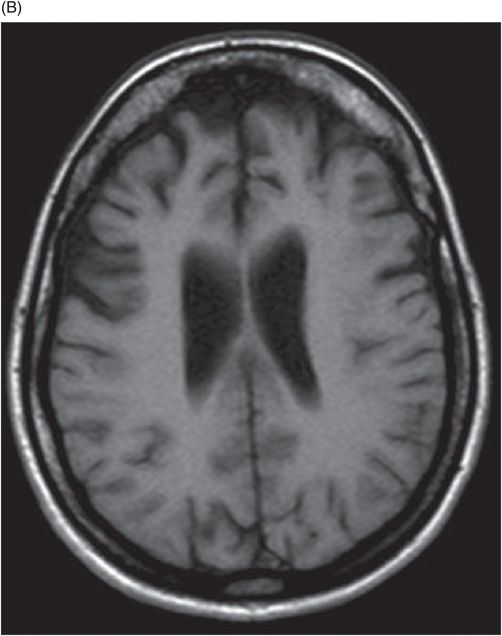
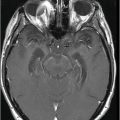
(A) Axial T1WI of the brain at the level of lateral ventricles and (B) Axial T1WI of the brain at the level of corona radiata.
Machado-Joseph Disease
Primary Diagnosis
Machado-Joseph disease
Differential Diagnoses
Sporadic olivopontocerebellar atrophy
Dentatorubral-pallidoluysian atrophy
Spinocerebellar ataxia type-6
Fragile X-associated tremor/ataxia syndrome
Cerebrotendinous xanthomatosis
Imaging Findings
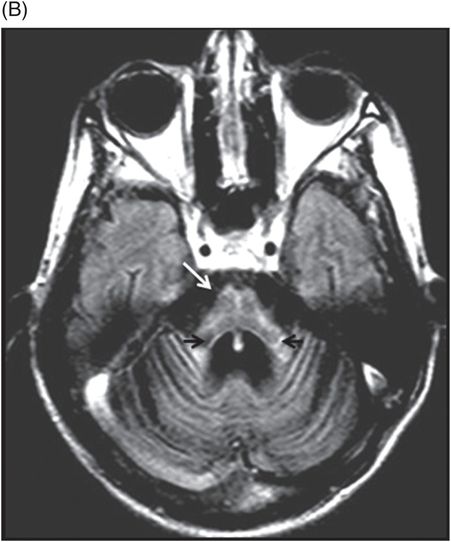
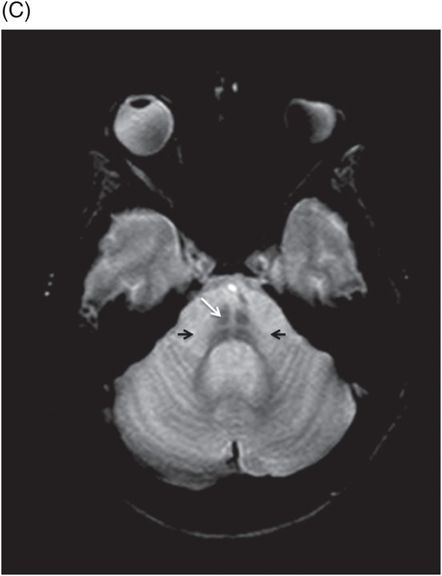
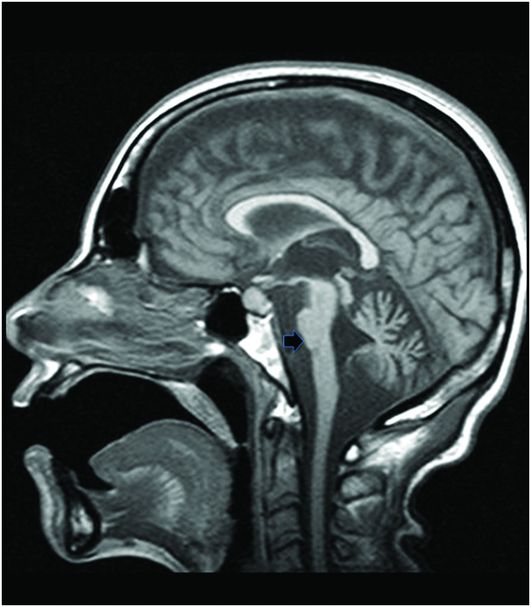
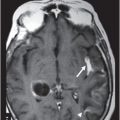
Fig. 20.1: (A) Axial T2WI through the brainstem and cerebellum demonstrated abnormal hyperintense signal changes in the pons giving the appearance of hot-cross-bun sign. (B) Axial FLAIR and (C) Axial GRE sequence also demonstrated hot-cross-bun sign (white arrow). Note marked atrophy of the dentate nuclei and middle cerebellar peduncles (black arrows) with significant cerebellar volume loss. Fig. 20.2: Sagittal T1WI showed pontine tegmental volume loss with flattening (arrow). Fig. 20.3: Coronal T2WI showed thinning of the superior cerebellar peduncles (arrows). Fig. 20.4: (A–B) Axial T1WI demonstrated generalized atrophy of both cerebral hemispheres.
Discussion
The imaging findings in this patient’s case overlap with multiple neurodegenerative conditions such as sporadic olivopontocerebellar atrophy (sOPCA), dentatorubral-pallidoluysian atrophy (DRPLA), and spinocerebellar ataxia type-6 (SCA type-6). Sporadic olivopontocerebellar atrophy, a condition with a similar but more rapid clinical course, usually spares the dentate nuclei, red nuclei, and superior cerebellar peduncles, frequently involved in patients with Machado-Joseph disease (MJD). Thus, a diagnosis of MJD is more likely based on the characteristic imaging features.
Other spinocerebellar neurodegenerative conditions such as DRPLA and SCA type-6 with similar involvement of the dentatorubral system and pallidosubthalamic system can be differentiated from MJD by the lack of ophthalmoplegia and atrophy of the facial nerve colliculus. Abnormal signal changes detected in the thalamus, brainstem, and cerebral white matter in DRPLA may be helpful in differentiating DRPLA from MJD. Fragile X-associated tremor/ataxia syndrome (FXTAS) is a sex-linked genetic disease with a male preponderance and middle-age onset (50 years of age) characterized by neuroimaging findings of atrophy and T2-weighted hyperintensity in the cerebellar hemispheres close to dentate nuclei and middle cerebellar peduncles. FXTAS demonstrates diffuse white matter hyperintensity – distinguishing it from MJD. Cerebrotendinous xanthomatosis (CTX) is an inherited metabolic condition resulting from the deposition of cholesterol and cholestanol in the nervous system. Early-onset clinical manifestations of CTX include low intellectual performance, ataxia, spastic paraparesis, peripheral neuropathy, and tendon xanthomas. Unique neuroimaging features of CTX patients such as T1 hyperintensity/T2-weighted hypointensity in the dentate nucleus (hyperdense on CT) differentiate it from MJD; thus, CTX can be excluded as a potential diagnosis in this case.
Machado-Joseph disease, also known as spinocerebellar ataxia type-3, is a hereditary autosomal dominant neurodegenerative disease that involves multiple systems. It was first described by Nakano et al. in a Portuguese-American family, who descended from Guilherme Machado, and emigrated from the Azores Island to Massachusetts. Since then, it has been described in other global populations. It is the most common and familial type of spinocerebellar ataxia caused by the expansion of CAG repeats in the MJD1 gene, now officially called ATXN3 gene located on chromosome 14q32.1. The disease is manifested in affected individuals between the ages of 25 and 55 years with a mean age of 40 years. The common clinical manifestations include progressive ataxia, ophthalmoplegia, extrapyramidal signs (dystonia and Parkinsonism), peripheral neuropathy, and other non-motor symptoms. Neuropathologically, selective brain areas including the dentate nucleus of the cerebellum, the nucleus dorsalis of Clarke in the spinal cord, cranial motor nerve nuclei, pontine nuclei, and substantia nigra are reported to show degeneration.
The most striking imaging features demonstrated in patients with MJD are moderate cerebellar atrophy and marked brainstem atrophy, especially in the tegmentum region of the pons. Other common imaging findings include atrophy of dentate nuclei, middle, and superior cerebellar peduncles with marked dilatation of the fourth ventricle, which can be explained by the atrophy of the brainstem and involvement of the afferent cerebellar tracts from the brainstem to cerebellum. Flattening of the facial colliculus is another finding that has been described in the literature. The non-specific T2-weighted high signal intensity in the transverse pontine fibers (hot-cross-bun sign) observed in various other degenerative cerebellar diseases, especially multiple system atrophy (MSA) (see Part I: Cases 8 and 9), also is described in MJD. There can be associated moderate to severe atrophy of the frontal and temporal lobes as well as marked atrophy of the globus pallidi, which correlates with the severity, and duration of the disease. Various SPECT and PET studies have demonstrated hypoperfusion and hypometabolism in the cerebral cortices involving the frontal, lateral temporal, parietal, and occipital lobes.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


