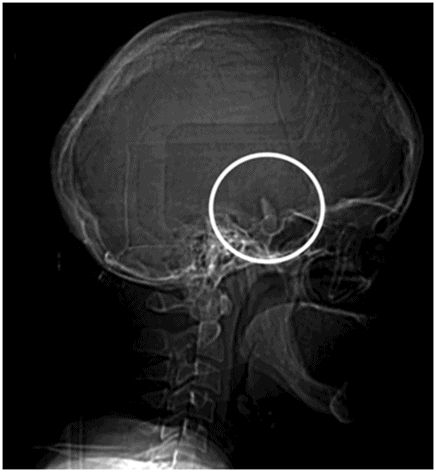
CT scanogram of the skull.
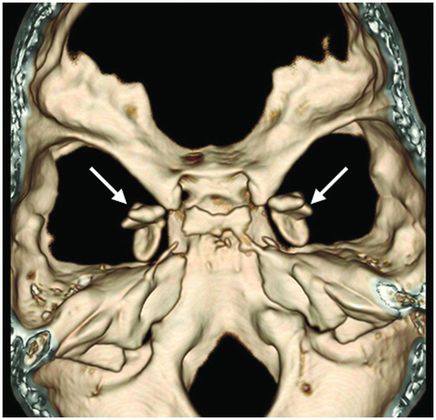
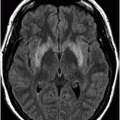
Three-dimensional volume rendering reformation of the skull base.
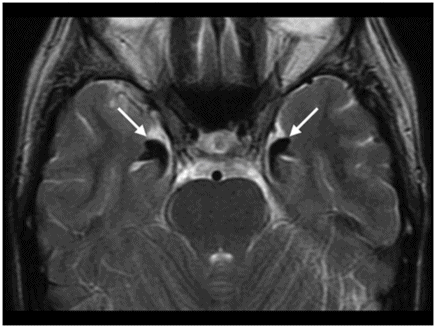
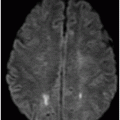
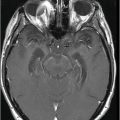
Axial T2WI at the level of the mesial temporal lobes.
Lipoid Proteinosis
Primary Diagnosis
Lipoid proteinosis
Differential Diagnoses
Ganglioglioma
Dysembryoplastic neuroepithelial tumor
Oligodendroglioma
Cavernous angioma (cavernoma)
Imaging Findings
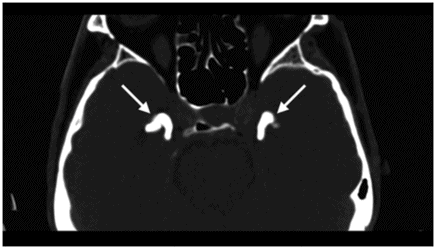
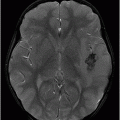
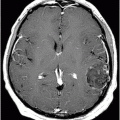
Fig. 61.1: Papular lesions in the eyelids, paucity of hair follicles in the eyebrow (black arrow), and complete absence of eye lashes (white arrow). Fig. 61.2: CT scanogram showed oval-shaped calcification projected at the sellar region (circle). Fig. 61.3: Three-dimensional volume rendering reformation from a volume CD acquisition showed bilateral symmetric triangular-shaped bilateral calcifications adjacent to the cavernous sinuses (arrows). Fig. 61.4: CT image at the level of the cavernous sinuses showed bilateral and symmetric, curvilinear comma-shaped calcifications at both mesial temporal lobes (arrows). Fig. 61.5: Axial T2WI at the level of the mesial temporal lobes showed bilateral, symmetric T2 hypointense curvilinear structures with no mass effect or edema in both unci (arrows).
Discussion
The long history of hoarseness, skin changes, and imaging findings are nearly pathognomonic for lipoid proteinosis.
Ganglioglioma is a benign neoplasm that commonly involves the temporal lobes. Although its lesions commonly calcify and present enhancement after contrast administration, they are rarely bilateral or present with a comma-shaped configuration. Dysembryoplastic neuroepithelial tumor is also a benign neoplasm typically located in the temporal lobes. Comparatively, its lesions show calcification less frequently than gangliogliomas and less commonly present contrast enhancement. Oligodendroglioma tumors show calcification in approximately 70% of the patients but are known to present enhancement.
Patients with the above-mentioned neoplasms can present with convulsions, particularly if their lesions are located in the temporal lobes. However, none of these lesions is associated with hoarseness or eyelid changes, or presents with bilateral symmetric calcification, calcification without mass effect, or adjacent parenchymal abnormalities. Bilateral comma-shaped calcification involving the amygdala, without enhancement or adjacent parenchymal abnormalities, is virtually pathognomonic of lipoid proteinosis.
Lipoid proteinosis (also known as hyalinosis cutis et mucosae or Urbach-Wiethe disease) is a rare autosomal recessive disorder characterized by generalized intracellular deposition of amorphous hyaline material, causing thickening and scarring of the skin, vocal cords, eyelids, mucosa, and viscera. The lungs, lymph nodes, striated muscles, and CNS may also be involved. Lipoid proteinosis was first described in 1908 by Siebenmann and further described in detail by Urbach and Wiethe in 1929.
Clinically, the most significant characteristics are hoarseness and moniliform blepharitis. Infantile hoarseness is a common presenting feature of the disease, due to infiltration of larynx, particularly the vocal cords. In two-thirds of the cases, voice changes are present at birth or in early infancy as the first manifestation. Moniliform blepharitis is characterized by nodules and papules in the eyelids with a beaded appearance that is pathognomonic of this condition. Other clinical features include cutaneous changes including waxy-textured skin, yellow papules, generalized skin nodules, and thickening. Hyperkeratosis may appear in regions exposed to mechanical friction, such as the hands, elbows, buttocks, axillae, and knees. The skin can be damaged by minor trauma or friction, resulting in blisters and scar formation. Scalp involvement may lead to hair loss and areas of alopecia. The mucosa of the pharynx, tongue, and soft palate may be hypertrophied and patients may suffer from dyspnea and difficulties in speech, requiring tracheotomy in severe cases.
The responsible gene involved in lipoid proteinosis encodes the extracellular matrix protein 1 (ECM1) on band 1q21. Patients with exon 7 mutations display slightly milder clinical features, while those with mutations on exon 6 manifest a severe phenotype. Polymerase chain amplification and direct nucleotide sequencing of the ECM1 gene can confirm the diagnosis.
The neuroimaging hallmark of lipoid proteinosis is the presence of CNS calcifications, secondary to glycoprotein material deposition in small arteries and vessels, especially in long-term patients. Calcifications mostly occur in the amygdala, hippocampus, parahippocampal gyri, and the striatum. Because of the temporal lobe calcifications, some patients develop epilepsy. The pathognomonic pattern is bilateral, symmetric, comma-shaped calcifications in the amygdala. In spite of being easily depicted, not all patients with lipoid proteinosis will show CNS involvement.
Computed tomography is an excellent imaging modality to show clearly the calcifications, their distribution, and pattern. On MRI, the most useful sequences are T2 gradient echo (T2*) and SWI. Usually, the calcifications tend to have low signal intensity in all sequences. Moreover, MRI is useful to rule out other differential diagnoses. Usually, symptoms improve over time. Nevertheless, voice alterations, speech difficulties, and airway obstructions tend to worsen, and older patients may need surgical procedures to maintain unobstructed airways.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


