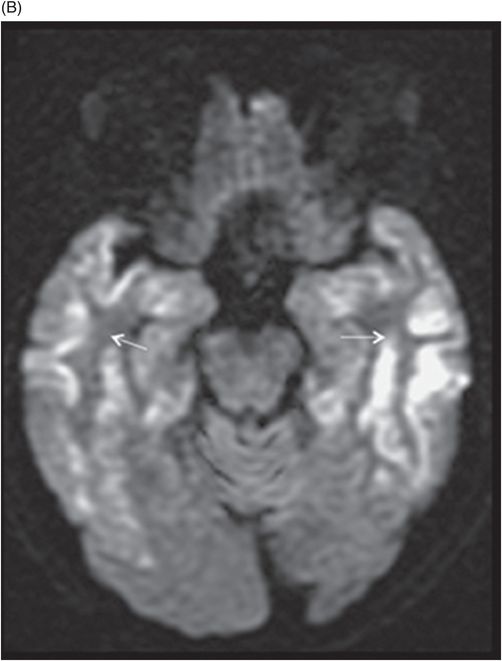
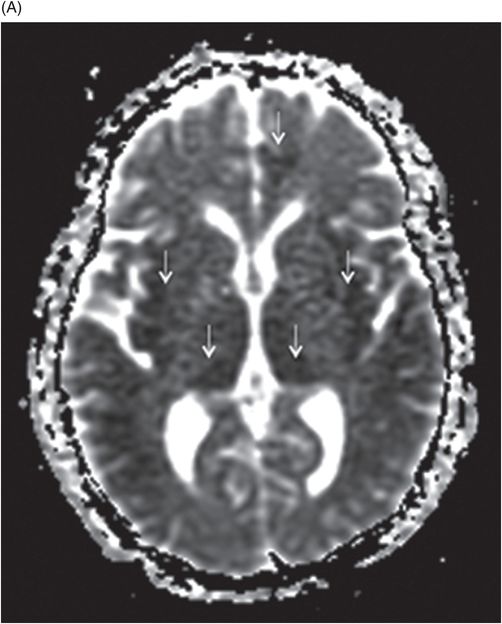
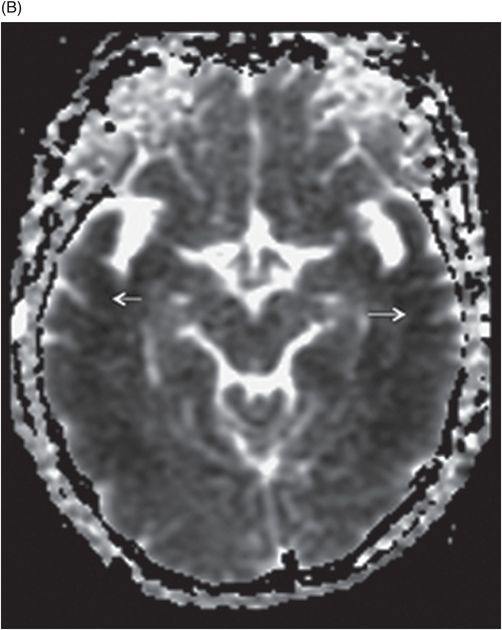
(A–B) Axial ADC maps through the same level.
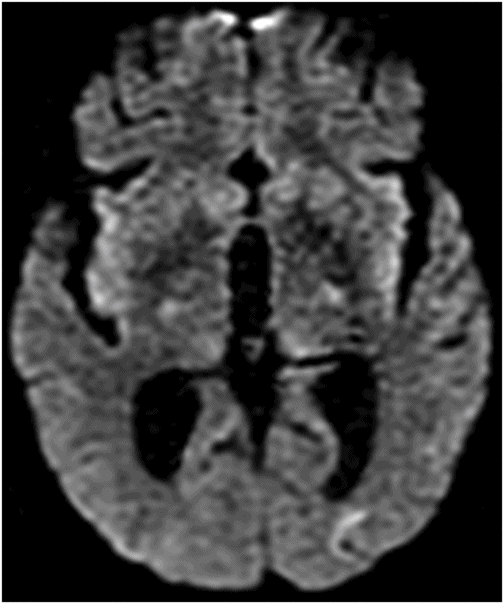
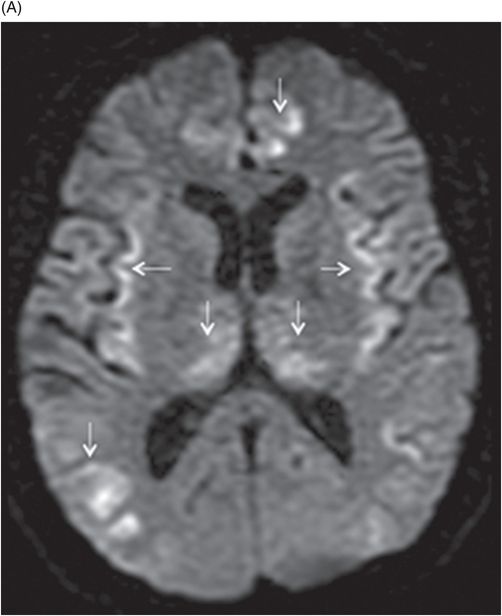
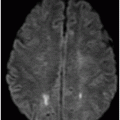
Axial DWI through the level of insular cortex and lateral ventricles.
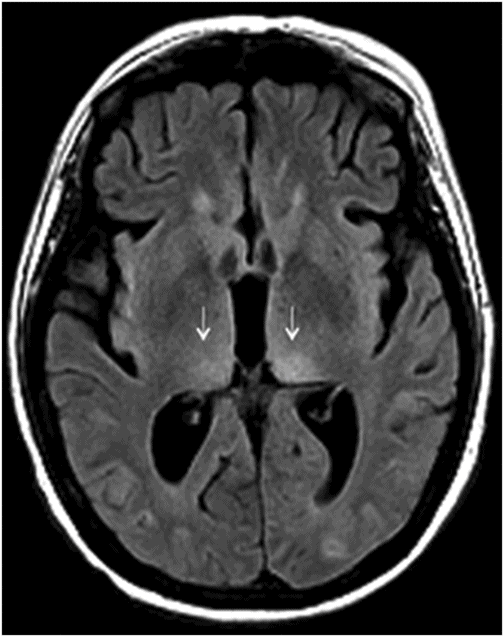
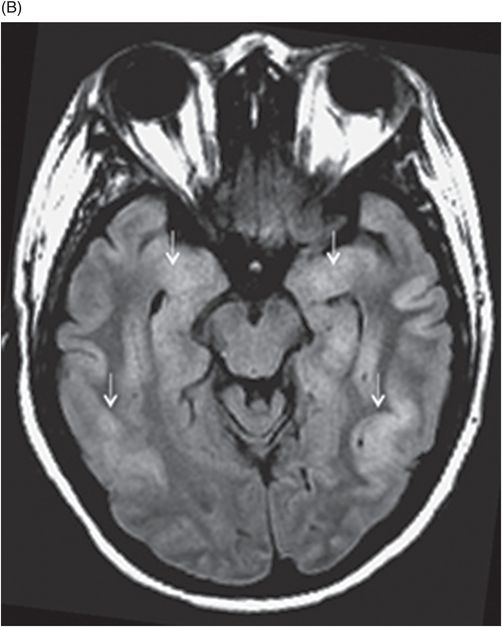
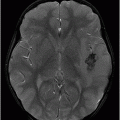
Axial FLAIR through the level of insular cortex, basal ganglia, and thalami.
Acute Hyperammonemic Encephalopathy
Primary Diagnosis
Acute hyperammonemic encephalopathy
Differential Diagnoses
Posterior reversible encephalopathy syndrome (PRES)
Paraneoplastic (autoimmune)/infectious encephalitis
Creutzfeldt-Jakob disease
Hypoxic ischemic injury
Imaging Findings
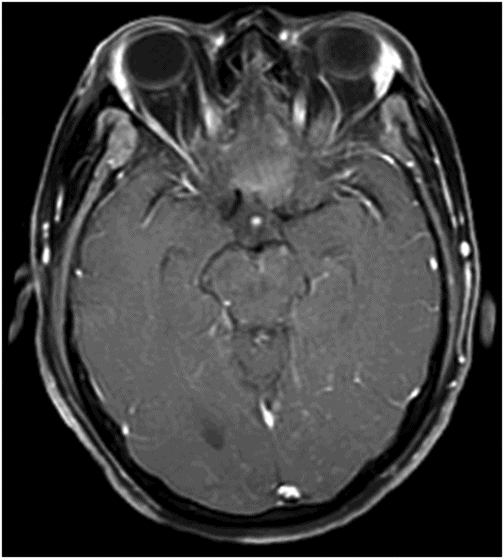
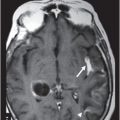
Fig. 68.1: (A–B) Axial FLAIR images of the brain showed hyperintense signal changes in the bilateral insular and cingulate cortices, thalami, and frontotemporal cortices (arrows). Fig. 68.2: (A–B) Axial DWI demonstrated restriction in the areas of hyperintensity noted on Fig. 68.1. Fig. 68.3: (A–B) Corresponding axial ADC map demonstrates changes. Fig. 68.4: Axial postgadolinium image did not show abnormal enhancement. Fig. 68.5: Axial follow-up DWI showed improvement in diffusion restriction signal changes. Fig. 68.6: Axial FLAIR image showed volume loss and residual FLAIR hyperintense signal abnormality; especially the thalami.
Discussion
Altered mental status, abnormal liver enzyme and ammonia levels, with involvement of the insular, cingulate, bilateral frontotemporal cortices, and thalami, with relative sparing of the basal ganglia on MRI are suggestive of hyperammonemic encephalopathy. Although imaging findings of hyperammonemic encephalopathy are well described in pediatric imaging literature, adult incidences are less prevalent. However, similar to this patient, brain MR images of affected individuals often demonstrate symmetric high signal on T2-weighted and FLAIR sequences, with diffusion restriction in the cingulate gyrus and insular cortex, sparing the perirolandic and occipital cortices. The cause of the cingulate gyrus and insular cortex susceptibility has yet to be elucidated. The perirolandic and occipital cortices are relatively resistant to injury, with no clear explanation.
Hyperammonemic encephalopathy is a potentially reversible condition, if the specific signal changes are identified at early symptom onset. High signal on T2-weighted and FLAIR sequences involving the subcortical white matter, basal ganglia, thalami, and brainstem can occur in severe cases. Pseudo-normalization changes on DWI and ADC occur approximately eight days after onset. Elevated glutamine peak can be seen on MR spectroscopy and correlates with the increased levels on CSF analysis. Later in the disease process, diffuse frontotemporoparietal cortical atrophy and high T1 signal intensity in the temporal cortex can be seen, indicating cortical laminar necrosis, a known consequence of hypoxic encephalopathy. The distribution of the abnormal cortical signal can be used to distinguish between hypoxic encephalopathy and hyperammonemic encephalopathy. The cortical laminar necrosis of hypoxic encephalopathy has a predilection for watershed areas and/or the parieto-occipital regions, whereas cortical laminar necrosis in hyperammonemic encephalopathy mainly involves the insular, cingulate, and frontotemporal cortex. High T1 signal of the basal ganglia is prominent, which may be attributable to underlying chronic liver disease or prolonged ammonia toxicity.
Seizure activity and diffuse hypoxic ischemic injury as potential diagnoses can be eliminated based on clinical history. Hematologic and CSF studies exclude paraneoplastic/infectious encephalitis, and Creutzfeldt-Jakob disease (CJD). On imaging, CJD also tends to involve the cingulate and frontal cortices. However, the lack of basal ganglia involvement, which is typical for CJD, and the acute onset of disease, does not correlate with the natural course of CJD. In addition, CSF analysis was negative for prion disease. Severe forms of PRES tend to involve the cortex and can show diffusion restriction, but the absence of white matter changes and lack of obvious precipitating events make it an unlikely cause.
Hyperammonemic encephalopathy can occur because of urea cycle defects, severe liver diseases, portosystemic shunting, sodium valproate or L-asparaginase therapy, infections, hypothyroidism, multiple myeloma, and in solid organ transplantations.
Ammonia is mainly produced in the gastrointestinal tract and is metabolized to form urea. The liver is the main source for ammonia metabolism. Disruption of normal liver function results in accumulation of ammonia, which is then metabolized by the kidneys, brain, and skeletal muscle. The increased level of ammonia in the CNS is directly proportional to systemic hyperammonemia. Imaging and clinical symptoms can be seen at blood ammonia levels ≥ 60 μmol/l and in the form of lethargy, irritability, vomiting, and somnolence. Rapid synthesis of glutamine from ammonia and glutamate through glutamine synthetase leads to astrocyte swelling, oxidative and nitrosative damage, defective neurotransmitter synthesis, disruption of glucose metabolism, and increased blood-brain barrier permeability. Hyperammonemia eventually affects multiple brain neurotransmitters, especially affecting glutaminergic NMDA receptors and modulating GABA receptors. It has been shown that glutamine levels correlate with encephalopathy severity. The metabolic disturbance of ammonia and glutamine leads to cytotoxic brain edema, intracranial hypertension, cerebral hypoperfusion, and decreased consciousness. Patients usually present with progressive drowsiness, seizures, and often become comatose because of the increasing effect of ammonia toxicity on the brain. Prolonged ammonia exposure can lead to serious brain damage, including intellectual impairment and irreversible sequelae.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


