Sonography is performed to confirm the presence of a clinically suspected mass and its site of origin. After the presence of an abdominal mass has been confirmed by sonography, patients undergo further imaging with either CT or MRI. Positron emission tomography (PET) and metaiodobenzylguanidine (MIBG) scintigraphy are also used in the staging of neuroblastoma.
Neuroblastoma
Neuroblastoma is the most common solid, extracranial malignant tumor in children, accounting for 8% to 10% of all childhood cancers (
4,
5,
6). Most children with neuroblastoma are between 1 and 5 years of age, with a median age at diagnosis of 22 months. Neuroblastomas may arise in the adrenal medulla or anywhere along the sympathetic ganglion chain. Up to 75% of neuroblastomas arise in the abdomen, and two-thirds of these lesions arise in the adrenal medulla. The remaining abdominal and pelvic tumors usually originate in the paravertebral sympathetic ganglia or presacral area. Occasionally, an abdominal tumor arises in the organ of Zuckerkandl. On cut-section, neuroblastoma is usually well demarcated, although it lacks a capsule, and it commonly contains areas of necrosis, hemorrhage, cystic degeneration, and calcification. Microscopically, the tumor is composed of small, darkly straining, round cells that are characteristically arranged in rosettes. Average tumor size is 8 cm at the time of presentation.
The most common clinical presentation of abdominal neuroblastomas is an abdominal mass. Other presenting features include skeletal pain; neurologic abnormalities, such as nystagmus, ataxia, opsoclonus, and paraparesis; ophthalmic signs, including proptosis and periorbital ecchymoses; and gastrointestinal complaints, such as diarrhea, anorexia, and vomiting. These signs and symptoms usually are related to metastatic spread of the tumor, intraspinal invasion, or the production of various hormones. Approximately 90% of patients with neuroblastoma have increased serum or urinary levels of catecholamines or their metabolites, particularly vanillylmandelic acid (VMA) and homovanillic acid (HVA). Between 60% and 70% of children have metastatic disease at time of diagnosis. Common sites of metastases are distant lymph nodes, cortical bone, bone marrow, liver, and skin. Neuroblastoma has been associated with neurofibromatosis type I and with aganglionosis of the colon.
Two paraneoplastic syndromes have been described in association with neuroblastoma. One is myoclonic encephalopathy of infancy, which includes the triad of cerebellar ataxia, myoclonus, and opsomyoclonus. The precise cause of this syndrome is unknown, but it has been postulated that an immune response to the primary tumor leads to production of antineural antibodies that cross-react with cerebellar tissue. The second syndrome consists of intractable watery diarrhea and hypokalemia caused by excess production of vasoactive intestinal peptides.
The International Neuroblastoma Staging System (INSS), based on the combination of clinical, radiographic, and surgical findings, is the system used most often to stage neuroblastoma (see
Table 9.1). Distribution of stages based the INSS is approximately I, 20%; II, 10%; III, 15%; IV, 50%; and IV-S, 4% (
5).
The treatment of neuroblastoma varies with the stage of the tumor. Patients with tumors that are localized to one side of the midline or cross the midline without encasement of major vessels undergo primary surgical resection. Chemotherapy is the treatment of choice in patients with unresectable disease. The use of radiation therapy is limited to patients with localized disease that cannot be resected and does not regress completely with chemotherapy. Once unresectable tumors decrease sufficiently in volume, delayed surgical resection is performed. Infants with IV-S disease may not be treated, as spontaneous regression sometimes occurs in these patients.
The principal determinants of prognosis are the stage and site of the tumor and the age of the patient. Tumors with lower stages, those arising at extra-abdominal sites, and tumors occurring in children under 1 year of age have a more favorable prognosis. Stage is a particularly important factor. Two-year survival ranges from 80% with disease limited to the adrenal to less than 5% for disease with skeletal metastases. Thoracic tumors also have a better overall survival rate than abdominal tumors. Several biological markers of tumor tissue also influence survival rate. Favorable factors include triploid karyotypes, well-differentiated stroma, an unamplified N-myc oncogene, and absence of abnormalities of chromosome 1. Unfavorable prognostic signs are a “diploid” (decreased DNA) karyotype, N-myc amplification (more than 10 copy numbers), and allelic loss of chromosome 1p.
Imaging Evaluation
Plain Radiography.
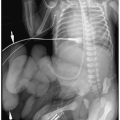
Plain films may be acquired for unrelated clinical indications and demonstrate an unsuspected primary tumor in the chest or abdomen or they may be obtained to further evaluate a suspected or known abdominal mass (most often diagnosed by sonography). Plain radiographic findings of abdominal neuroblastoma include a paraspinal mass and enlargement of the intervertebral foramina or erosion of the pedicles due to intraspinal extension of tumor. The tumor may contain calcifications.
Sonography.
The evaluation of patients with palpable abdominal masses, including neuroblastoma, usually begins with sonography (
1,
6,
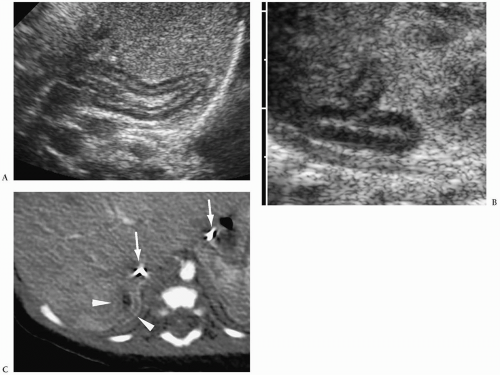
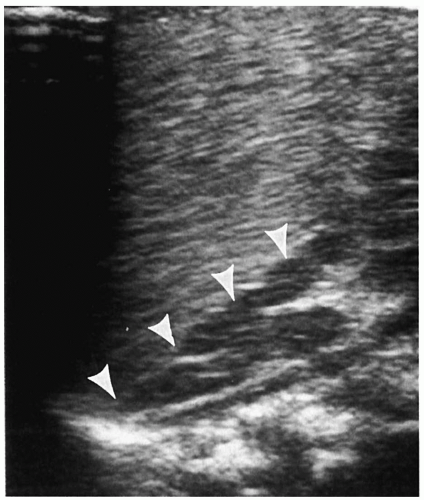
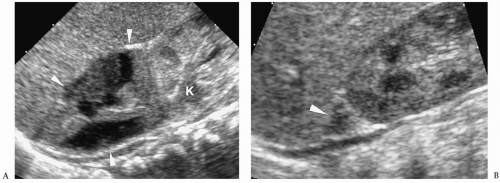
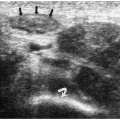
7). Neuroblastoma appears as a suprarenal or paraspinal mass. It may be homogeneous or heterogeneous and it may contain hyperechoic areas representing calcification and hypoechoic areas as a result of hemorrhage, necrosis, or cystic degeneration (
Fig. 9.4). Tumor margins may be smooth or irregular. Peripheral or central vascularity may be observed on color Doppler sonography.
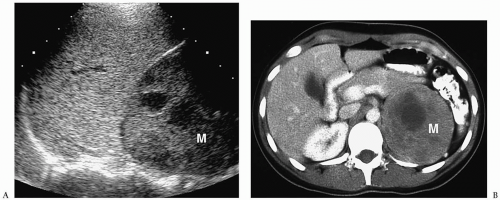
In the newborn, neuroblastomas may be predominantly cystic or anechoic (
8,
9) (
Fig. 9.5). On pathologic sections, this appearance reflects either degenerative change in the tumor or, in some cases, clusters of microcysts in the tumor cells.
Computed Tomography.
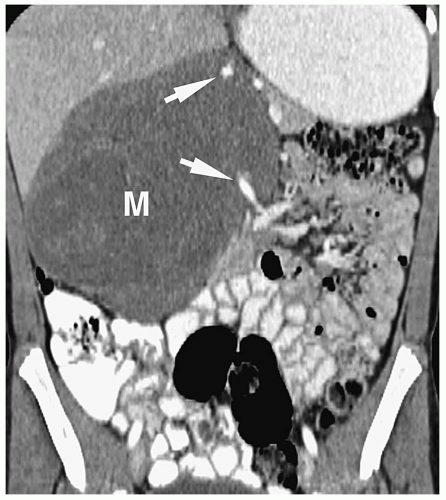
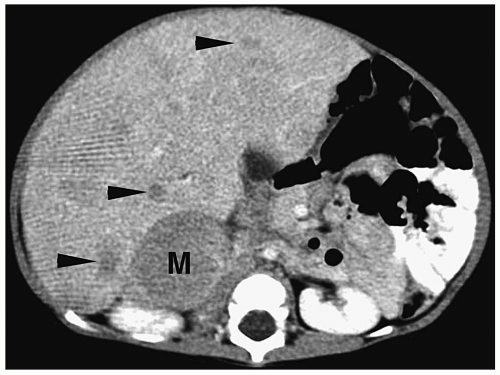
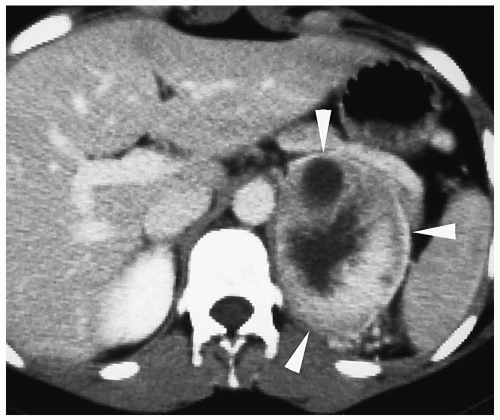
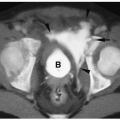
On CT, neuroblastoma appears as a homogeneous or heterogeneous, soft tissue mass with irregular margins (
2,
4,
6,
7). The tumor enhances less than that of surrounding tissues after intravenous administration of contrast material. Calcifications occur in approximately 85% of abdominal neuroblastomas (
Fig. 9.6).
Magnetic Resonance Imaging.
Neuroblastoma has low or intermediate signal intensity on T1-weighted sequences and high signal intensity on T2-weighted and fat-suppressed sequences (
4,
6,
10,
11). It usually enhances after administration of intravenous gadolinium chelate compounds (
Fig. 9.7). Calcification has low signal intensity on all imaging sequences. Areas of necrosis and cyst formation are usually hypointense on T1-weighted images and hyperintense on T2-weighted sequences and do not enhance following gadolinium administration.
Imaging Intra-abdominal Tumor Spread.
Patterns of intra-abdominal extension of tumors include midline extension, vessel encasement, spread to regional lymph nodes, intraspinal extension, and hepatic metastases. Inferior vena caval and aortic displacement are common. Bone metastases also may be seen.
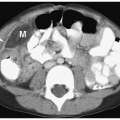
Hepatic metastases have a variable appearance. The lesions can be discrete and solitary or multifocal. Discrete hepatic lesions are hyperechoic or hypoechoic on sonography, low attenuation on CT, hypointense to adjacent liver on T1-weighted MR images, and hyperintense on T2-weighted and fat-suppressed images. Diffuse metastatic infiltration produces hepatomegaly with heterogeneity of the hepatic parenchyma (
Fig. 9.8).
Imaging Distant Tumor Spread.
Distant spread is most commonly to lymph nodes and skeleton. A common site of distant nodal spread is the supraclavicular region.
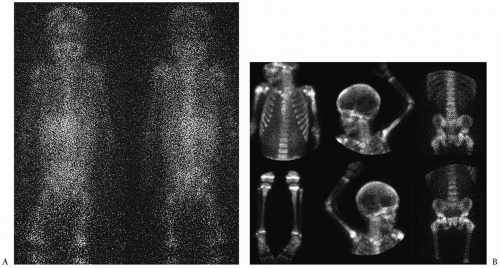
Skeletal metastases occur in 50% to 60% of patients at diagnosis, mostly in patients older than 1 year of age (
5). These can involve cortical bone or bone marrow. MIBG has become the study of choice to image skeletal metastases (
12) (
Fig. 9.9). Abnormal activity can be seen in the primary tumor and in bone, bone marrow and soft tissue metastases. PET performed with the glucose analogue 2-[fluorine-18]-fluoro-2-deoxy-D-glucose (FDG) has been used most recently for the evaluation of distant disease extent. Most neuroblastomas and their metastases avidly concentrate FDG prior to chemotherapy or radiation therapy (
13). Skeletal metastases also can be seen on MRI, where they have a low signal intensity on T1-weighted sequences and high signal intensity on T2-weighted and fat-suppressed sequences.
Follow-up Imaging.
CT, MRI, MIBG, and PET have been used to assess the response of the tumor to surgery, chemotherapy, and radiation therapy. Neuroblastoma tends to regress in size with treatment, although the regression may be incomplete, leaving a residual retroperitoneal soft-tissue mass.
Pheochromocytoma
Pheochromocytoma is a functionally active neoplasm of the adrenal medulla that causes catecholamine overproduction. Most pheochromocytomas originate in the adrenal medulla, but up to 30% may arise in extraadrenal sites, mainly the paravertebral sympathetic chain, paraaortic bodies, and bladder wall. When this tumor originates outside of the adrenal glands, it is termed a paraganglioma. Most pheochromocytomas in children are benign.
Clinical features suggestive of the diagnosis are paroxysmal hypertensive episodes, headache, tachycardia, palpitation, diaphoresis, and pallor. Patients with bladder wall paragangliomas may present with micturition syncope. There is an increased incidence of pheochromocytomas in patients with multiple endocrine neoplasia type 2 (medullary thyroid carcinoma and parathyroid disease), and in patients with neurofibromatosis and von Hippel-Lindau disease. Multiple or bilateral tumors are more likely in these syndromes. The diagnosis of pheochromocytoma is usually established by biochemical testing, which demonstrates elevated urinary or serum catecholamines levels or elevated urine metanephrine or vanillylmandelic acid levels.
Localization of pheochromocytomas is accomplished with CT, MRI, or 123I- or 131I-labeled MIBG. Total body scanning with MIBG is especially useful in patients with multifocal tumors or metastatic disease.
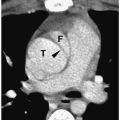
Pheochromocytomas are usually large (>2 cm in diameter), sharply marginated lesions (
1,
2,
3,
4,
11) (
Fig. 9.10). Smaller tumors commonly have a homogeneous matrix, whereas larger tumors are often heterogeneous. Calcifications are present in about 15% of tumors. The tumors are predominantly soft-tissue attenuation on CT, hypointense to liver on T1-weighted images, and markedly hyperintense to liver and fat on T2-weighted and fat-suppressed images. Moderate to marked heterogenous enhancement after intravenous administration of contrast medium or gadolinium is typical. Signs of malignancy, such as local invasion, lymph node enlargement and distant metastases, may also be seen. The imaging appearance of pheochromocytoma is not specific and differentiation from other adrenal tumors requires biochemical studies.