Fig. 4.1
Schematic representation of the main molecular mechanisms involved in the antisecretory effects of somatostatin analogs. cAMP cyclic adenosine monophosphate; Gi G inhibitory protein; SST somatostatin; PKA protein kinase A
Indications
Octreotide is approved for the control of hormone-related symptoms in NETs in the U.S. and in Europe, and lanreotide is approved for the same indication in Europe only. Since their introduction in routine clinical practice in the 1980–1990s, SST analogs have clearly improved therapeutic management of patients with functional NETs [17]. The first clinical trial with octreotide, published in 1986, involved 25 patients with carcinoid syndrome [18]. Flushing and diarrhea were promptly relieved in 22 patients (88 %), and 18 (72 %) had a decrease of ≥50 % in their urinary 5-HIAA levels compared with pretreatment values, leading to approval of octreotide by the Food and Drug Administration (FDA). Subsequently, about 30 studies were conducted with SST analogs, involving more than 500 patients, most of whom had carcinoid syndrome (review in [3]). The mean symptom control rate was 73.2 %, and no difference was found between the various agents and formulations. SST analogs appeared more effective against flushing than diarrhea. In a study published in 2004 by Ruszniewski et al. [19], 71 patients with carcinoid syndrome were enrolled to receive lanreotide Autogel® at a dose of 90 mg every 4 weeks for two injections, after which the dose was adapted over 6 months according to the patient’s response. Flushing episodes and diarrhea were significantly decreased at 6 months compared with baseline (p <0.001 and p = 0.006, respectively), and biochemical response (decrease of ≥ 50 % in serum chromogranin A and/or urinary 5-HIAA levels) was observed in 30 % of patients. In a crossover study comparing octreotide with lanreotide in 33 patients with carcinoid syndrome, O’Toole et al. [20] demonstrated equivalent efficacy for the two compounds in terms of symptom control and reduction in tumor markers. Pooled data analysis from 15 studies confirmed that octreotide LAR® and lanreotide Autogel® yield similar symptom control rates of 74.2 % (95 % confidence interval [CI], 61.9–92.8 %) and 67.5 % (95 % CI, 40.0–100 %), respectively, as well as for biochemical response rates (51.4 % [95 % CI, 31.5–100 %] and 39.0 % [95 % CI, 17.9–58 %], respectively) (Fig. 4.2, part 1) [3]. These findings were also reported in acute situations, such as during carcinoid crisis, which may occur spontaneously or during tumor manipulation in surgical or radiologic procedures [21]. A phase III study of pasireotide LAR® versus octreotide LAR® in metastatic NET patients with functional symptoms inadequately controlled by SST analogs was presented at the ASCO 2013 meeting (abstract #4031, NCT00690430). A total of 110 patients have been randomized and were stratified by predominant symptom at baseline (diarrhea, flushing, or both). Symptom response rates at 6 months were not significantly different between pasireotide LAR® and octreotide LAR® (21 vs. 27 %; odds ratio [OR] = 0.73, p = 0.53).
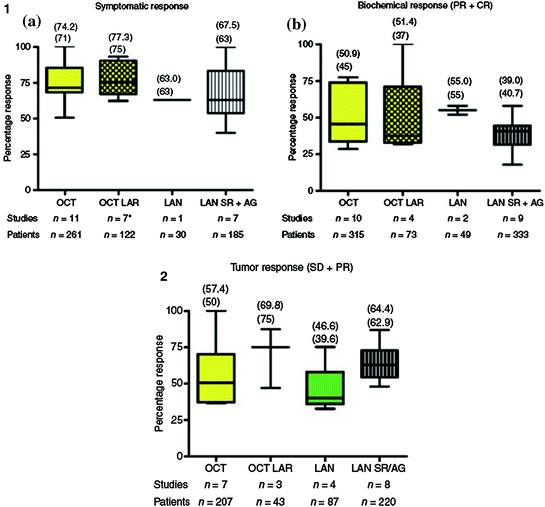
Fig. 4.2
Efficacy of different somatostatin analogs and formulations in terms of antisecretory effects and antitumor effects (data compilation from 15 studies, in [3]). OCT octreotide; OCT LAR octreotide LAR®; LAN lanreotide; LAN SR/AG lanreotide Autogel®; PR partial response; CR complete response; SD stable disease Mean (top) and median in brackets. (1) Antisecretory effects, (2) Antitumor effects
SST analogs are also effective against other NET hormone-related symptoms (review in [22, 23]). While insulinomas and gastrinomas are the most frequent functional pancreatic NETs, use of SST analogs in these indications is limited [9, 17]. In gastrinomas, high doses of proton-pump inhibitors are essential to control Zollinger–Ellison syndrome caused by gastrin hypersecretion, and the use of SST analogs is limited to rare refractory cases. In insulinomas, the effect of SST analogs on glycemia is unpredictable and hypoglycemia may be either improved or worsened by SST analogs due to glucagon suppression, requiring careful management of this treatment with initiation during hospitalization. Moreover, other treatments are available and effective in controlling insulin hypersecretion: diazoxide, which inhibits insulin release by direct action on β cells, or more recently, everolimus [24]. In contrast, the beneficial role of SST analogs has been well documented in less common types of functional pancreatic NETs [9, 17]. In glucagonomas, SST analogs are effective in controlling necrolytic migratory erythema in 80–90 % of patients, although their effect on the associated diabetes and weight loss is less pronounced [9, 17]. In VIPomas, diarrhea and electrolyte imbalance, also known as Werner–Morrisson syndrome or WDHA (watery diarrhea, hypokalemia, achlorhydria) syndrome, are significantly improved by SST analogs in 80–90 % of patients, although their effect may be limited by tachyphylaxis [9, 17].
Antitumor Effects
Recently, SST analogs were demonstrated to exert antitumor effects in selected NET patients and underlying molecular mechanisms have been elucidated.
Molecular Basis
The molecular mechanisms responsible for the antitumor effects of SST analogs are classified as direct and indirect mechanisms [7]. Direct mechanisms are associated with cell cycle arrest and/or apoptosis downstream of SSTR activation. Mechanisms inhibiting cancer cell proliferation are complex and involve several intracellular signaling pathways which depend on the SSTR subtype (Fig. 4.3). All SSTRs induce expression of cell cycle inhibitors, including p27 and p21, leading to cell cycle arrest at the G1/2 (SSTR1, 2, 4, and 5) or G2/M (SSTR3) phase [7, 16]. SSTR1, 2, 3, and 4 trigger the recruitment and activation of phosphotyrosine phosphatases (PTPs) in a Gi/Go-dependent manner [7]. These PTPs (SHP-1, SHP-2, PTPη) subsequently dephosphorylate growth-factor-bound tyrosine kinase receptors (TKRs) and phosphorylated tyrosine residues of TKR targets (e.g., c-Src), thereby inhibiting growth factor signaling and modulating downstream effectors such as mitogen-activated protein (MAP) kinases (SSTR1, 2, 3, 4), PI3 K/AKT (SSTR2), and nitric oxide (NO) pathways (SSTR1, 2, 3) [7, 16]. SSTR5, on the other hand, does not require PTP to exert its antiproliferative effect. It is mediated by Gi/Gq-dependent inhibition of: (1) phospholipase C (PLC) and IP3 which regulate Ca2+ influx, (2) cGMP and downstream MAP kinase signaling, and (3) the src-like tyrosine kinase p60Src that inactivates NO synthase [7]. Moreover, SSTR2 and SSTR3 were also shown to induce apoptosis through p53-dependent (SSTR3) or p53-independent (SSTR2) mechanisms [7, 16]. Both apoptotic pathways are affected by SST, the extrinsic pathway, through sensitization to death receptors of TNF-α, TRAIL, and Fas-Ligand (SSTR2 and SSTR3), and the intrinsic (mitochondrial) pathway, through inhibition of anti-apoptotic proteins such as Bcl-2 (SSTR2) and induction of pro-apoptotic proteins such as Bax (SSTR3) [7, 16]. The apoptotic effect of SSTR2 also involves the inhibition of survival signals mediated by MAP kinases and PI3 K/AKT pathways [7, 16].
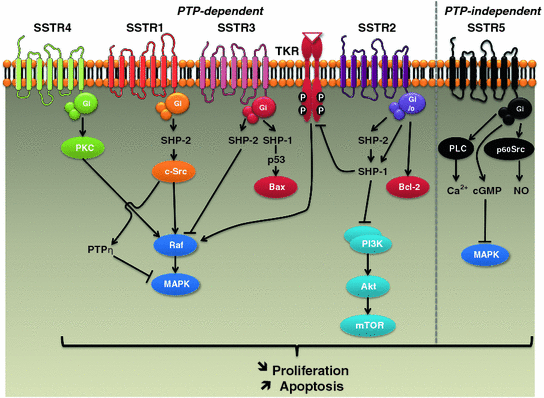
Fig. 4.3
Schematic representation of main molecular mechanisms involved in antitumor effects of somatostatin analogs. MAPK mitogen-activated protein kinase; PKC protein kinase C; PLC phospholipase C; PTP phosphotyrosine phosphatase; SSTR somatostatin receptor; TKR: tyrosine kinase receptor
Overall, growth inhibition is mediated mainly by SSTR2 and SSTR5, while apoptosis is triggered by SSTR3, which may explain why octreotide and lanreotide, targeting mainly SSTR2 and SSTR5, often display cytostatic rather than cytotoxic effects. In addition, SST also displays anti-invasive properties in certain tumor types. This effect may be due to SSTR1, 2, 3, and 4-mediated inhibition of the small GTPases Rac or Rho, both of which regulate cytoskeleton organization and cell migration [7, 16].
Indirect mechanisms mainly involve SST-induced inhibition of growth factor secretion and tumor angiogenesis, and modulation of immune cells [7, 16]. SST impacts the growth hormone (GH)/insulin growth factor (IGF)-1 axis both centrally, through inhibition of GH synthesis (SSTR2 and SSTR5), and peripherally, through down-regulation of STAT5b-mediated IGF-1 gene transcription in the liver (SSTR2 and SSTR3) [7, 16]. In addition, endothelial cells express SSTR1, 2, 3 and 5, and SST decreases their proliferation and migration [7, 16]. Inhibition of endothelial NO synthase and expression of proangiogenic factors such as VEGF, PDGF, or bFGF, also contributes to the antiangiogenic effect of SST [7, 16].
Defining the Indications: The PROMID and CLARINET Studies
Tumor responses were reported early in the clinical development of SST analogs, and a large number of clinical trials were initiated to investigate the antitumor effect of these agents, although the underlying molecular mechanisms were not yet clearly understood (Fig. 4.2, part 2) [17]. In a systematic review of 28 prospective studies (17 octreotide trials and 11 lanreotide trials) published from 1989–2011, partial response (PR) rates ranged from 0–31 % and stable disease (SD) from 15–89 % [25].
The PROMID study was the first phase III trial to demonstrate that long-term administration of SST analogs can control tumor growth in NET patients [26]. In this study, 85 treatment-naïve midgut NET patients were randomly assigned to receive either monthly octreotide LAR® 30 mg (n = 42) or placebo (n = 43) until tumor progression or death. The majority (61 % of patients) had nonfunctioning tumors. Most patients had tumors with a Ki67 proliferation index less than 2 % (95 % of patients) and were octreoscan-positive (74 % of patients). Median time to progression (TTP) in the octreotide LAR® arm was 14.3 months compared with 6.0 months in the placebo arm (hazard ratio [HR] = 0.34, p = 0.000072). After 6 months of treatment, SD was observed in 66.7 % of patients in the octreotide LAR® arm versus 37.2 % in the placebo arm (p = 0.0079). The greatest benefit was observed in patients with low hepatic tumor burden (≤10 %) and resected primary tumor. The effect was independent of tumor functionality. The study was not designed to demonstrate superiority in terms of overall survival since more than 90 % of patients with tumor progression in the placebo arm received octreotide LAR. Based on the results of this study, the National Comprehensive Cancer Network (NCCN) guidelines were revised to include the use of SST analogs as antineoplastic agent for metastatic midgut NETs.
However, many criticisms emerged subsequent to the publication of this study. The major reproach was the lack of progression status prior to inclusion. Since evidence of progressive disease (PD) was not a requirement for study entry, the indication for starting active treatment with an SST analog in otherwise asymptomatic patients is unclear [27]. Moreover, there was a significant imbalance in the time since diagnosis between the study arms in favor of the octreotide LAR® arm, with a median of 7.5 months compared with 3.3 months for the placebo arm (p = 0.0096). Longer time between diagnosis and treatment suggests a more indolent disease [27]. Finally, these results left partially unanswered the question of which patients are most likely to benefit from SST analog treatment, since the benefit was more important in, but probably not limited to, patients with metastatic midgut NETs with Ki67 proliferation index ≤2 %, hepatic tumor burden ≤10 % and resected primary tumor. In a retrospective study conducted in 68 patients with metastatic digestive NETs, pretreatment stability (HR = 0.241, p = 0.008), Ki67 proliferation index ≤5 % (HR = 0.262, p = 0.009), and hepatic tumor burden ≤25 % (HR = 0.237, p = 0.004) were significantly associated with SD under lanreotide therapy in multivariate analysis, suggesting that PROMID-derived criteria could be expanded [28].
More recently, the results of the CLARINET study were presented at the ESMO 2013 meeting (abstract #E17-7103, NCT00353496). CLARINET is a phase III trial evaluating the antiproliferative effects of lanreotide Autogel® in patients with advanced gastroenteropancreatic NETs. A total of 204 patients with well or moderately differentiated (Ki67 proliferation index <10 %), octreoscan-positive, nonfunctioning NETs who had not received SST analogs or other treatment in the prior 6 months were enrolled. They were randomized to receive lanreotide Autogel® 120 mg (n = 101) or placebo (n = 103) every 4 weeks for 96 weeks, or until tumor progression or death. Primary tumor locations were pancreatic (45 %), midgut (36 %), hindgut (7 %), and unknown (13 %). Most patients were treatment-naïve (81 %) and had SD (96 %) prior to inclusion. Twenty-two percent of patients had WHO grade 2 tumors (Ki67 = 3–10 %), and 33 % had a hepatic tumor burden of more than 25 %.
After two years of treatment, median progression-free survival (PFS) was 18.0 months in the placebo arm and had not been reached in the lanreotide Autogel® arm (HR = 0.47, p = 0.0002), while 22 % of placebo subjects were alive compared with 62 % in the lanreotide group (HR = 0.47, p = 0.0002). In subgroup analysis, the increase in PFS with lanreotide Autogel® was significant compared with placebo for patients with midgut tumors (HR = 0.35, p = 0.0091), but not for patients with pancreatic tumors (HR = 0.58, p = 0.0637). Benefit was similar for WHO grade 1 and grade 2 tumors and was greater for patients with a lower hepatic tumor burden (≤25 %, HR = 0.34, p = 0.0002) but remained significant in patients with a greater hepatic tumor burden (>25 %, HR = 0.45, p = 0.0170). The results of this study contribute to expand and better define the indications of SST analogs in digestive NETs.
Data for pasireotide are scarcer. In the phase III study evaluating symptomatic effect of pasireotide LAR® versus octreotide LAR® presented at the ASCO 2013 meeting (abstract #4031, NCT00690430), patients on pasireotide LAR® had a 5 month longer PFS than patients on octreotide LAR® (investigator assessment), despite no differences in symptom response rates. These results warrant a large phase III trial to clarify the antitumor role of pasireotide.
Current Limits and Perspectives with SST Analogs
Given their mechanisms of action, SST analogs can be considered targeted agents directed against SSTR. As for all targeted therapies, their use in the clinic is limited by the emergence of acquired resistance, also known as tachyphylaxis. Furthermore, in some NET patients, SST analogs lose effectiveness within months of treatment initiation, whereas in other patients, tumor symptoms and growth can be controlled for several years. The reasons for tachyphylaxis are unclear but may be due to reduced SSTR expression on NET cells, activation of alternative pathways, and/or changes in SST analog pharmacokinetics when chronically administered. Three major strategies have been developed to enhance SST analogs efficacy and/or overcome acquired resistance: dose optimization, combination with other agents, and new SSTR-binding molecules.
Dose Optimization
In some patients, increasing the dose may restore the original response [17]. In clinical practice, this may be achieved either by a higher dose or by shortening the interval between injections (review in [29]). In a retrospective 8-year study in 108 patients with metastatic midgut NETs with carcinoid syndrome, 24 % had a sustained symptomatic response [30]. In the remaining patients, loss of symptomatic response with the initial dose was noted within 3–60 months. In 17 % of them, symptoms were controlled by an increase in octreotide LAR® dose, while the other patients required additional treatment.
The highest approved dose of octreotide LAR® is 30 mg administered every 4 weeks. A retrospective study of digestive NET patients requiring higher (40–90 mg) doses to control symptoms refractory to conventional doses of octreotide showed that dose escalation was safe [31]. In the dose-titration study of lanreotide Autogel®, 45 (63 %) of the 71 patients were treated with 120 mg/4 weeks, 11 (16 %) with 90 mg/4 weeks (initial dose), and 15 (21 %) with 60 mg/4 weeks. Twenty-seven (38 %) responded to doses of 120 mg or less, 15 (21 %) to 90 mg or less, and 11 (15 %) to 60 mg [19]. Dose optimization was well tolerated and caused a reduction in episodes of flushing and diarrhea by a mean of 1.3 and 1.1 episodes/day, respectively (p < 0.001). Welin et al. [32] evaluated the effect of a high-dose octreotide regimen (octreotide 160 mg/2 weeks) in 12 patients with progressive advanced midgut carcinoid tumors. Ten patients had symptomatic improvement of flush and diarrhea, and tumor size and biochemical markers were stabilized for a median of 12 months in 75 % of the patients. The results of the HIDONET trial (NCT00990535) evaluating a more frequent dosing schedule of octreotide LAR® (30 mg/3 weeks) in patients with progressive NETs are pending.
In addition, a study reported a decrease of 50–70 % of SST analogs plasma levels over a 2 year period in patients chronically treated with SST analogs [33]. This may be explained by either: (1) diminished bioavailability of the agent due to granulomatous reaction at the injection site, (2) development of antibodies against the agent, or (3) altered pharmacokinetics and metabolism of the agent due to changes in disease status or the patient (such as body mass index) [17]. However, the usefulness of plasma SST analog concentrations for treatment optimization is not clearly established, and plasma concentration monitoring during chronic SST analog therapy is not recommended [17].
Combination Therapy
The cytostatic rather than cytotoxic activity of SST analogs, thus inhibiting proliferation rather than inducing apoptosis, opens the opportunity for combination therapy with other agents, either chemotherapy or targeted agents. In vitro and in vivo data using SSTR2-positive colon cancer cell lines showed that SST analogs may have an additive/synergistic effect when combined with chemotherapy, such as 5-FU or mitomycin C [17]. Adding SST analogs potentiated the antiproliferative effect (S-phase block) and increased apoptosis of cancer cells. SST analogs can also be combined with targeted agents. Recently, phase III trials have demonstrated that targeted therapies directed against receptors of VEGF (sunitinib) or mTOR (everolimus) produced clinically significant improvement in patients with digestive NETs [34, 35].
SST analogs and everolimus both act on the PI3 K/AKT/mTOR signaling cascade, controlling protein synthesis and cell survival. Synergistic effects are exerted by enhancing signal inhibition on the downstream target 4E-BP1 [16]. SST analogs (through their action on endothelial cells and inhibition of VEGF production) and everolimus (through inhibition of the mTOR-HIFα axis) may also synergistically decrease angiogenesis. Finally, by reducing IGF-1 levels, SST analogs may overcome resistance from everolimus-induced regulation of IGF-1 pathway. The RADIANT-1 open-label phase II study assessed the clinical activity of everolimus (10 mg/day), with or without octreotide LAR® (30 mg/4 weeks) based on prior octreotide therapy, in patients with pancreatic NETs, after failure of chemotherapy [36]. Median PFS was 16.7 and 9.7 months with combination therapy and monotherapy, respectively. Subsequently, the RADIANT-2 phase III study compared everolimus (10 mg/day) plus octreotide LAR® (30 mg/4 weeks) to placebo plus octreotide LAR® in 429 patients with progressive low- or intermediate-grade advanced NETs [37]. Median PFS was significantly improved in the everolimus plus octreotide LAR® group (16.4 months vs. 11.3 months in the placebo plus octreotide LAR® group, HR = 0.77, p = 0.026). Drug-related adverse events were mostly grade 1 or 2, including stomatitis, rash, fatigue, and diarrhea. Phase I (NCT00804336, NCT01263353, NCT01590199) and II (NCT01374451) studies of pasireotide LAR® in combination with everolimus in digestive NETs are currently ongoing.
There is also a rationale for combining SST analogs with antiangiogenics. Well-differentiated NETs are remarkably highly vascularized and display sensitivity to intra-arterial (chemo) embolization and antiangiogenic agents (see Chap. 11). Combination of SST analogs with anti-VEGF antibodies (e.g., bevacizumab) or tyrosine kinase inhibitors (e.g., sunitinib) may yield enhanced antiangiogenic and antitumoral effects. Combination of octreotide LAR® (30 mg/4 weeks) with temozolomide (100 mg/day) and bevacizumab (7.5 mg/m2/3 weeks) in 15 patients with advanced NETs, mainly WHO grade 2, showed interesting activity, with tumor control in 13 (86 %) patients (1 complete response, 8 PR, 3 SD) and a median TTP of 36 weeks [38]. A phase II study compared combinations of octreotide LAR® (continued at pre-study dose) with either bevacizumab (15 mg/m2/3 weeks) or pegylated interferon α-2b or in patients with advanced extrapancreatic NETs [39]. Bevacizumab plus octreotide LAR® resulted in a high tumor control rate (95 %, including 18 % PR and 77 % SD), significant decrease in tumor blood flow (p < 0.01) and longer PFS (18 week PFS rate of 95 vs. 68 % in the interferon plus octreotide arm). The results of the subsequent phase III study (NCT00569127) are pending, along with the phase II study of combined bevacizumab, pertuzumab (HER1/HER2 inhibitor) and octreotide LAR® for advanced NETs (NCT01121939), and the phase II study of everolimus and octreotide LAR® with or without bevacizumab for advanced pancreatic NETs (NCT01229943). However, these studies were not designed to assess the benefit of combination therapy versus SST analog monotherapy. The SUNLAND phase II study, which evaluates sunitinib versus placebo in combination with lanreotide Autogel® in patients with progressive advanced midgut NETs (NCT01731925), is ongoing.
To summarize, there is emerging evidence for additive/synergistic activity of SST analogs combined with targeted agents. Such combinations may be of particular interest in patients with NETs progressing under SST analogs. However, the underlying mechanisms are not clearly defined and appropriate clinical studies are required to specifically assess the effect of combination therapy versus SST analog monotherapy.
New SSTR-binding Molecules
Advances in the understanding of SSTR signaling, trafficking, and interactions have led to the development of new SSTR-binding molecules. Besides pan-SSTR analogs such as pasireotide, compounds cotargeting SSTRs and other receptors have emerged as a promising strategy. The observation that the majority of well-differentiated NETs coexpress SSTRs and the dopamine type 2 receptor (D2R) and that SSTR and D2R can form heterodimers with enhanced functional activity, provided a rationale for the development of new chimeric compounds that can bind both receptor types [40–42]. BIM-23A760 was the first molecule of this family. It was designed to have high affinity for SSTR2 and D2R, while its affinity for SSTR5 was intentionally low to reduce the risk of hyperglycemia. Phase II clinical studies have been performed in patients with carcinoid syndrome (NCT01018953) and acromegaly (NCT00994214) [9]. However, its clinical development has been stopped and new chimeric molecules are currently being developed. Among them, BIM-23A758 (which is an SSTR2/D2R chimeric compound as well) induced significant antitumor effects in human GOT1 midgut carcinoid cells and may be a promising new molecule for NET therapy [43].
Peptide-receptor-targeted Radiotherapy
Peptide-receptor scintigraphy such as octreoscan is useful in NET imaging for diagnosis and staging (see Chap. 2). Moreover, it can be used to select patients for the therapeutic strategy using radiolabeled SST analogs, PRRT [44]. Different radiolabeled SST analogs have been used, all of which share a common structure, comprising three parts: the radionuclide itself (Indium-111 [111In], Yttrium 90 [90Y], or Lutetium 177 [177Lu]), a chelator (diethylene triamine penta-acetic acid [DOTA] or 1,4,7,10-tetra-azacyclododecane-1,4,7,20-tetra-acetic acid [DTPA]), and an SST analog (octreotide or lanreotide). Modifications in the radionuclide and chelator can considerably affect compound characteristics, including pharmakocinetics and SSTR-binding affinity [45].
Early studies were performed with high doses of 111In-DTPA0-octreotide, the same radiolabeled peptide as is used for octreoscan imaging. 111In is a γ-emitter creating Auger electrons with short range and low tissue penetration (review in [46]), which may only be effective for the treatment of micrometastatic or low-burden disease, and is unsuitable for treating bulky tumors. This may explain the disappointing results of 111In-coupled peptides in clinical studies in metastatic NET patients, with low rates of objective tumor response (0–17 %) compared with 90Y- or 177Lu-coupled peptides. 90Y- or 177Lu are β-emitter with higher energy and tissue penetration (reviewed in [47] and [48], respectively). 90Y-DOTA-octreotide (DOTATOC) and 177Lu-DOTA-octreotate (DOTATATE) are the two most widely used radiopeptides for PRRT in metastatic NETs, both yielding similar response rates (15–35 %) [45, 49]. More than 500 patients have been treated with 177Lu-DOTATATE and more than 300 with 90Y-DOTATOC in clinical studies, respectively. Results are summarized in Table 4.1. Differing antitumor effects between studies may be due to different administered doses and dosing schemes, total tumor burden including the extent of liver involvement, and patient characteristics.
Table 4.1
 New Anticancer Agents in Neuroendocrine Tumors
Inhibition of mTOR in Neuroendocrine Neoplasms of the Digestive Tract
Clinical Management of Targeted Therapies in Neuroendocrine Tumours
New Anticancer Agents in Neuroendocrine Tumors
Inhibition of mTOR in Neuroendocrine Neoplasms of the Digestive Tract
Clinical Management of Targeted Therapies in Neuroendocrine Tumours
 Overcoming Resistance to Targeted Therapies: The Next Challenge in Pancreatic Neuroendocrine Tumors (PNETs) Treatment
Overcoming Resistance to Targeted Therapies: The Next Challenge in Pancreatic Neuroendocrine Tumors (PNETs) Treatment
 Measuring the Relationship of Quality of Life and Health Status: Including Tumor Burden, Symptoms, and Biochemical Measures in Patients with Neuroendocrine Tumors
Measuring the Relationship of Quality of Life and Health Status: Including Tumor Burden, Symptoms, and Biochemical Measures in Patients with Neuroendocrine Tumors
 Imaging of Neuroendocrine Tumors and Challenges in Response Evaluation for Targeted Therapies
Imaging of Neuroendocrine Tumors and Challenges in Response Evaluation for Targeted Therapies




Summary of main clinical studies of peptide-receptor radionuclide therapy (PRRT) with 90Y- or 177Lu-labeled somatostatin analogs in patients with digestive neuroendocrine tumors (adapted from [45, 49])
Related posts:
New Anticancer Agents in Neuroendocrine Tumors
Inhibition of mTOR in Neuroendocrine Neoplasms of the Digestive Tract
Clinical Management of Targeted Therapies in Neuroendocrine Tumours
Overcoming Resistance to Targeted Therapies: The Next Challenge in Pancreatic Neuroendocrine Tumors (PNETs) Treatment
Measuring the Relationship of Quality of Life and Health Status: Including Tumor Burden, Symptoms, and Biochemical Measures in Patients with Neuroendocrine Tumors
Imaging of Neuroendocrine Tumors and Challenges in Response Evaluation for Targeted Therapies
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
