Fig. 8.1
Upstream activation of the mTOR signaling pathway and consequences of mTORC1 inhibition using everolimus in pancreatic neuroendocrine tumors
Illustrations
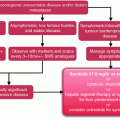
Genetic diseases associated with the development of digestive neuroendocrine tumors.
Hereditary forms of neuroendocrine tumors have highlighted the crucial role of genes regulating hypoxia signaling. In von Hippel–Lindau disease, loss of pVHL protein function, which usually tags the hypoxia-inducible factor 1 (HIF1) for proteasomal degradation, results in nuclear accumulation of HIF1α, yielding increased transcription of a number of hypoxia-inducible genes, such as CA-9 and GLUT-1 (glucose transporter 1). In type 1 neurofibromatosis and tuberous sclerosis syndromes, HIF-1α is indirectly activated through mTOR (mammalian target of rapamycin), due to loss of function of NF1 and TSC1 genes. Of note, such cases of familial pNET are acknowledged to be associated with adverse outcome as compared to MEN1 tumors.
References
1.
Halfdanarson TR, Rabe KG, Rubin J, Petersen GM (2008) Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol 19:1727–1733PubMedCentralPubMedCrossRef
2.
3.
Ballian N, Loeffler AG, Rajamanickam V, Norstedt PA, Weber SM, Cho CS (2009) A simplified prognostic system for resected pancreatic neuroendocrine neoplasms. HPB 11:422–428 (Oxford)PubMedCentralPubMedCrossRef
4.
5.
Modlin IM, Pavel M, Kidd M, Gustafsson BI (2010) Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine (carcinoid) tumours. Aliment Pharmacol Ther 31:169–188PubMed
6.
7.
8.
9.
Heng PN, Saltz LB (1999) Failure to confirm major objective antitumor activity for streptozocin and doxorubicin in the treatment of patients with advanced islet cell carcinoma. Cancer 86:944–948
Related posts:
 New Anticancer Agents in Neuroendocrine Tumors
Clinical Management of Targeted Therapies in Neuroendocrine Tumours
New Anticancer Agents in Neuroendocrine Tumors
Clinical Management of Targeted Therapies in Neuroendocrine Tumours
 Angiogenesis Inhibition Using Sunitinib in Pancreatic Neuroendocrine Tumors
Angiogenesis Inhibition Using Sunitinib in Pancreatic Neuroendocrine Tumors
 Overcoming Resistance to Targeted Therapies: The Next Challenge in Pancreatic Neuroendocrine Tumors (PNETs) Treatment
Overcoming Resistance to Targeted Therapies: The Next Challenge in Pancreatic Neuroendocrine Tumors (PNETs) Treatment
 Measuring the Relationship of Quality of Life and Health Status: Including Tumor Burden, Symptoms, and Biochemical Measures in Patients with Neuroendocrine Tumors
Measuring the Relationship of Quality of Life and Health Status: Including Tumor Burden, Symptoms, and Biochemical Measures in Patients with Neuroendocrine Tumors
 Imaging of Neuroendocrine Tumors and Challenges in Response Evaluation for Targeted Therapies
Imaging of Neuroendocrine Tumors and Challenges in Response Evaluation for Targeted Therapies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


