Fig. 1
a Left: DNA Binding Domains (DBD) of the heterodimer USP/EcR on the DNA target, an inverted repeat separated by one base (IR1). PDB ID: 2HAN. Right: DBDs of homodimer RXR. The DNA target is a direct repeat separated by one base (DR1) PDB ID: 4CN2. b DNA Binding Domains (DBD) on direct repeat separated by 1, 3, 4 and 5 bases (DR1, DR3, DR4 and DR5). PDB ID: 3DZU for PPAR/RXR DBD DR1. CryoEM structure for VDR/RXR DBD DR3. PDB: ID 4NQA for LXR/RXR DBD DR4. SAXS/SANS/FRET structure for RAR/RXR DBD DR5
4 LBDs Heterodimers
The LBD fold is constituted by a primarily helical scaffold termed “antiparallel α-helical sandwich” of 12 helices and a short β-turn arranged in three layers. This arrangement generates a mostly hydrophobic ligand-binding pocket (LBP) which can accommodate natural or synthetic ligands. The pocket can adapt to accommodate different ligands (Billas et al. 2003; Huet et al. 2015) . In the agonist-bound (holo) LBD the C-terminal helix H12 is stabilized against H3 and H11, thereby sealing the ligand-binding cavity. This conformation is specifically induced by the binding of agonists and is referred to as the “active conformation”. It favors the recruitment of transcriptional coactivators to the receptor surface composed of helices H3, H4 and H12.
The crystal structures of a limited number of unliganded (apo) LBDs (Bourguet et al. 1995) , together with biochemical data, have revealed that H12 either adopts a different position or exists as a dynamic ensemble of conformations (Nahoum et al. 2007; le Maire et al. 2010) . In both apo-RXR from human (hRXR) and the invertebrate chordate amphioxus (AmphiRXR), the LBPs are filled with hydrophobic residues of H11, thereby stabilizing the ligand-free cavity. Comparison of the apo- and holo-LBD structures of hRXRα illustrates the mechanism by which the activation function AF-2 becomes transcriptionally competent upon ligand binding (Egea et al. 2000, 2001) (Fig. 2). The structural transition essentially involves the stabilization of helix H11 in the continuity of helix H10, and the repositioning of helix H12 that seals the LBP and further stabilizes ligand binding.
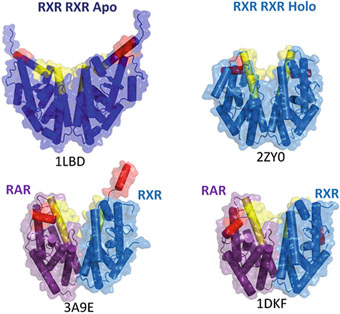
Fig. 2
Dimers of Ligand Binding Domains (LBD), the helices H11 and H12 are colored in yellow and red respectively. Top: left homodimer of apo-RXRs (without ligand) (1LBD), right: homodimer of liganded RXRs in the agonist conformation (2ZY0). Bottom: heterodimers of liganded RAR/RXR, RAR is bound to agonist ligands, whereas RXR is bound to an antagonist (3A9E, left) or an agonist ligand (1DKF, right)
Several structures of homo- and hetero-dimers, notably those of RXR LBD in complex with various partner LBDs, including RAR (Bourguet et al. 2000; Pogenberg et al. 2005; Sato et al. 2010) , the peroxisome proliferator activated receptor (Gampe et al. 2000) , the thyroid hormone receptor (Putcha et al. 2012) , the liver X receptor (Svensson et al. 2003) or the constitutive androstane receptor (Suino et al. 2004; Xu et al. 2004) , have been reported thereby identifying the structural organization of receptor dimers. All these structures demonstrate a topologically conserved dimerization surface with residues from helices H7, H9, H10, as well as loops L8–9 and L9–10 of each protomer forming a network of complementary hydrophobic and charged residues. Upon dimerization the hidden surface of each monomer is about 1000 Å2 (Table 1). Extensive analyses of the dimerization interfaces based on crystal structure and sequence alignment have provided insights for the specific dimerization characteristics of nuclear receptors (Bourguet et al. 2000; Brelivet et al. 2004) .
Table 1
Buried surface of representative homo and hetero dimers. (Binding area of 2 monomers)
PDB | Dimer | Surface dimer (A²) | Buried surface (A²) | |
|---|---|---|---|---|
Chain A | Chain B | Complex | Complex | |
1LBD | RXRα | RXRα | 23073,543 | 1836,076 |
2ZY0 | RXRα | RXRα | 21496,027 | 2187,918 |
3A9E | RXRα | RARα | 24607,131 | 1600,08 |
1DKF | RXRα | RARα | 21676,182 | 1881,687 |
5 DNA Binding Mechanism and Architecture of the Complexes
In solution RAR and RXR form stable elongated complexes with bound LBD heterodimers and DBDs loosely positioned through their flexible hinge domains (Fig. 3). Upon binding to target DNA, the heterodimer adopts an asymmetric conformation with two distinct modules, the DBDs bound to the DNA RE and the heterodimeric LBDs, positioned at the 5-end of the RE. The structures then suggest that DNA recognition is achieved by the preassembled heterodimer instead of the heterodimer being constituted on the DNA target. The process of a combined approach to the recognition of the response element is an efficient way to overcome the specificity problem with low affinity constants for each single DBD. It is tempting to generalize the mechanism to most NRs but more experimental data are needed.
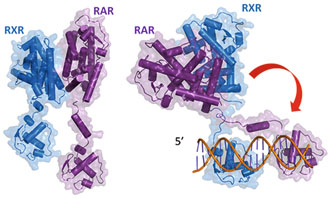
Fig. 3
Two solution structures of full length RAR/RXR in their free state (left) and bound to their target DNA (DR5) (right)
The structural data explain how DNA dictates the architecture of the complex and its asymmetry, and several pieces of evidence further support the proposal. Velasco et al. showed that the binding of the SRC-1 receptor interacting domain to thyroid hormone receptor (TR) is influenced by the DNA response elements (Velasco et al. 2007) . The thyroid response element organization dictates the composition of active receptor. An allosteric communication between SRC-1 and DNA was also reported for VDR-RXR (Zhang et al. 2011) .
The solution structures of full length or ΔAB RAR/RXR heterodimers free and bound to the best characterised RAREs (DR1 and DR5) together with that of PPARα/RXRα/DR1 and VDR/RXRα on its DR3 response element were first determined using integrative approaches (Rochel et al. 2011) . The molecular structures revealed some common features that suggest functional correlations that could be relevant for a large fraction of the superfamily. Regardless of the different positions and polarities of the bound DBDs the complexes exhibit a similar extended and asymmetric conformation with the LBD dimer positioned on the 5ʹ side of the response elements. In all cases the LBDs and the DBDs/DNA entities are nearly orthogonal, the pseudo two-fold symmetry axis of the LBDs and the DNA forming the two branches of an L-shaped structure. The LBDs dimers can rotate around both their pseudo two-fold axis and the DNA axis under the control of the flexible hinge domains. The position of the DBDs on the DNA response elements is dictated by the relative position of the binding motifs. For example addition of one base pair to the spacer sequence induces a shift of approximately 3.5 Å and a rotation of 36°. Thus for a given heterodimer the response elements control the architecture of the complexes through the polarity of the hexa-nucleotide binding motifs and the number of dinucleotide spacers. Together with the hinges they modulate the rotation of the LBDs and the relative position of the receptors. The hinge domains play an important role (see below).
The cryo-EM structure of the liganded human RXR and VDR bound to a consensus DNA response element forming a direct repeat (DR3) fits the experimental SAXS data and confirms the features of the solution structure (Orlov et al. 2012) . The LBDs are perpendicular to the DNA and are located asymmetrically at the DNA 5ʹ end of the response element. The hinges of both VDR and RXR are fully visible. They hold the complex in an open conformation. The asymmetric topology of the complex provides the structural basis for RXR being an adaptive partner within NR heterodimers while the helical structure of VDR’s hinge connects the 3ʹ-bound DBD with the 5ʹ bound LBD in a more constrained and specific manner (see below).
The cryo EM structure of the USP/EcR/IR1 complex provides the first description of a full length receptor bound to an inverted repeat (Fig. 4). USP/EcR binds to half-sites with a 1 base-pair spaced inverted repeat (IR1), a palindromic DNA response element reminiscent of IRs observed for vertebrate steroid hormone receptors (SHRs). The structure reveals that even though the DNA is almost symmetric, the complex adopts a highly asymmetric architecture in which the ligand-binding domains (LBDs) are positioned 5ʹ off-centered on the RE which leads to additional interactions between the USP LBD and the 5ʹ-flanking sequence that trigger transcription activity as monitored by transfection assays. With respect to an orientation perpendicular to the DNA, the LBDs are slightly tilted (~ 20°) towards the 5ʹ direction and rotated by ~ 30° around the pseudo-two-fold symmetry axis that goes through the LBD interface of USP and EcR. This results in an asymmetric arrangement where the USP LBD is closer to the DNA than the EcR LBD which leads to additional interactions between the USP LBD and the 5ʹ-flanking sequence that trigger transcription activity as monitored by transfection assays. Although the LBDs of USP and EcR are pseudo-symmetric they can be distinguished unambiguously in the cryo-EM map thanks to specific structural differences. Remarkably, compared to DR-bound NR complexes, the position of the LBDs is inverted in the IR1-bound complex, a topology which arises from the opposite orientation of the second RE half-site. The structure shows that helix H12 of EcR it is in the agonist position (due to presence of the ecdysteroid ligand) while that of USP adopts an antagonist conformation due to the presence of a phospholipid ligand (Billas et al. 2003) .
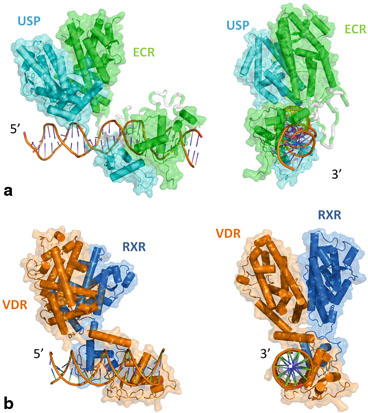
Fig. 4
a CryoEM structure of EcR/USP bound to the target DNA IR1 (inverted repeat separated by 1 bases). The hinge part is modeled (not present in the PDB: 4UMM). b Two orthogonal views of the cryo-EM structure of VDR/RXR bound to their target DNA (DR3)
6 The Hinge Domains
The architecture of the different complexes points to the important role of the hinge domains in establishing and/or maintaining the integrity of the functional structures. The Table 2 illustrates the large size fluctuation of these domains. The flexibility of the RXR hinge is a structural necessity to adapt to numerous partners and even more response elements with different polarities and spacers. Conversely the hinges of the partner receptors may have a more constrained structure in order to improve the specificity of DNA recognition while stabilizing the functional architecture. VDR and the thyroid hormone receptor (TR) provide a good example of these properties. The structure of VDR/RXRα/DR3 shows that the long and extended hinge domain of RXR is needed to reach helix H1 on the side opposite from the RXR DBD. For RXR a rigid C terminal helix similar to that of VDR positioned at the 5ʹ side of the response element would entirely change the topology of the complex.
Table 2
Hinge domain size
NR | Number of residues | Average |
|---|---|---|
VDR | 33 | 33 |
TR | 38 | 38 |
RAR | 25–26 | 25,11 |
LXR | 41–63 | 50,75 |
EcR | 71–93 | 79,57 |
FXR | 51–56 | 54,28 |
PPAR | 30–31 | 30,47 |
HNF4 | 17–20 | 17,37 |
RXR | 20–29 | 22,05 |
USP | 20–61 | 31 |
In the VDR/RXRα/DNA complex the helix orientation fits both the experimental molecular envelope and the distance requirements to join the N terminus of the LBD. A close, compact conformation for RXR–VDR would require the disruption of the helix. Mutation studies on VDR and RXR hinges (14 deletion mutants for VDR and 8 deletion mutants for RXR) fully support the solution structure, notably the requirement of the entire VDR hinge for full transcriptional activation (Shaffer et al. 2005) . Deletion of region 114–120 is deleterious, but the mutation of the residues into alanine does not affect transcription (Hsieh et al. 1999) . The role of this sequence-independent region is structural: it controls the relative spatial positions of the LBD and the DBD. By contrast, the mutation of residues 108–114 into alanine leads to a loss of transcription capability, underlying the critical role of these residues in positioning the LBD and DBD through contacts with the promoter. On the other hand up to 14 residues can be deleted from the more flexible hinge domain of RXR without marked effect on the transcription activity. This flexibility confers RXR plasticity and adaptability towards different response elements and heterodimeric partners. Interestingly in the crystal structures of DNA bound DBDs the α-helical hinge domains of VDR and TR exhibit similar orientations despite different crystal packing (Rastinejad et al. 1995; Shaffer and Gewirth 2002, 2004) . The sequence homology between VDR and TR hinges suggest a molecular model of TR/RXR/DR4 fitting all previously defined functional criteria (Fig. 5)
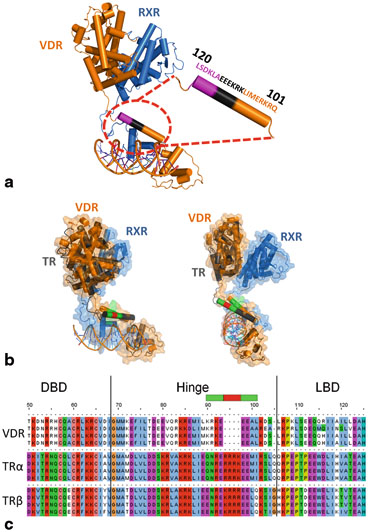
Fig. 5
 Multi-Functional Androgen Receptor Coregulator and Proto-Oncogene Melanoma Antigen-A11 (MAGE-A11)
Multi-Functional Androgen Receptor Coregulator and Proto-Oncogene Melanoma Antigen-A11 (MAGE-A11)
 Estrogen Receptor Modulators (SERMs) and Selective Androgen Receptor Modulators (SARMs)
Estrogen Receptor Modulators (SERMs) and Selective Androgen Receptor Modulators (SARMs)
 Analyses of Ordered and Disordered Regions in Ecdysteroid Receptor
Analyses of Ordered and Disordered Regions in Ecdysteroid Receptor
 and Regulation of Nuclear Receptor Corepressor Complexes
and Regulation of Nuclear Receptor Corepressor Complexes
 Receptor Structure and Function
Receptor Structure and Function
 Regulation and Intrinsic Disorder in Nuclear Hormone Receptors
Regulation and Intrinsic Disorder in Nuclear Hormone Receptors




a Solution structure of VDR/RXR/DNA. The zoomed area is the hinge part of VDR. The entire hinge is required for full transcriptional activity. In magenta? (114–120) it’s a sequence independent spacer. In black (108–114) it’s a sequence where mutations to alanine leads to a loss of transcriptional activity. b Two orthogonal views of the molecular models of VDR/RXR/DR3 (solution structure) and TR/RXR/DR4 (proposed model) superimposed. VDR is in orange and TR in grey. The larger hinge of TR allows to reach a LBD in a position close to that of VDR despite a longer distance (DR4). For TR model the crystal structures of DBD (PDB ID: 3M9E) and LBD (PDB ID: 3UVV) were used. For the hinge model the homologous amino acids between VDR and TR are shown in green, the insertion in the TR sequence is in red. c Sequence alignment of VDR and TR hinges. The green and red parts are correlated with the Fig. 5b
Related posts:
Multi-Functional Androgen Receptor Coregulator and Proto-Oncogene Melanoma Antigen-A11 (MAGE-A11)
Estrogen Receptor Modulators (SERMs) and Selective Androgen Receptor Modulators (SARMs)
Analyses of Ordered and Disordered Regions in Ecdysteroid Receptor
and Regulation of Nuclear Receptor Corepressor Complexes
Receptor Structure and Function
Regulation and Intrinsic Disorder in Nuclear Hormone Receptors
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
