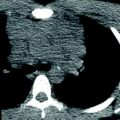
Fig. 1
Double aortic arch with dominant left arch in a neonate with severe respiratory distress. Posterior oblique (a) and posterior cranial (b) volume rendered, axial (c) and coronal (d) CT images show a complete vascular ring with a dominant left arch (green arrow), which is unusual, completely encircling the trachea (yellow arrow) and the esophagus (red arrow). The trachea is severely compressed at the level of the ring (white arrow in c). Coronal (e) and sagittal (f) minimum intensity projection images show pronounced hyperinflation, evidenced by flattening of the hemidiaphragms and bulging of lung tissue between the ribs (asterisks). Note the extent and severity of the tracheal compression (black arrow). The esophagus is air distended (white arrows) and is also collapsed, and likely compressed at the level of the ring (arrowhead). The narrowing is so severe that on virtual bronchoscopy (g) the trachea appears to end blindly (arrow)
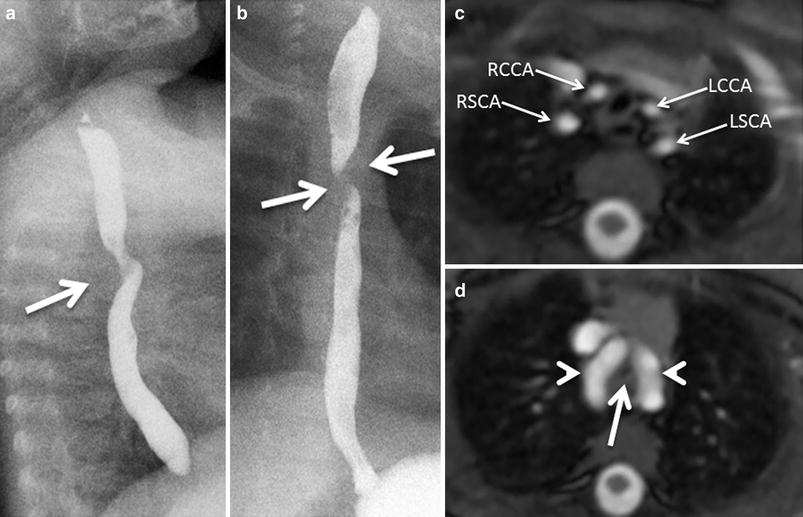
Fig. 2
A 6-month-old with worsening stridor and double aortic arch with equal arches. Lateral (a) and frontal (b) esophagogram images show broad posterior (a) and bilateral (b) indentations consistent with vascular impressions on the esophagus. c. Axial white-blood MR image shows symmetrical origins of the four arch vessels arising separately from the two arches. RCCA right common carotid artery; RSCA right subclavian artery; LCCA left common carotid artery; LSCA left subclavian artery. d. More inferior axial image reveals two nearly equal aortic arches (arrowheads) encircling the narrowed trachea (arrow). This type of double aortic arch, although it is not the more common type found in clinical practice, it is usually associated with significant tracheal compression
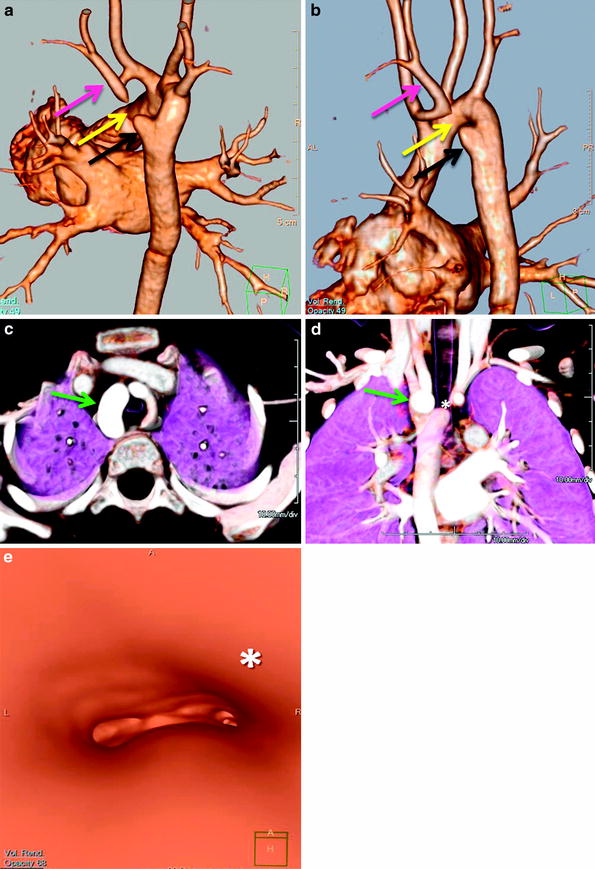
Fig. 3
Double aortic with atretic left arch. Posterior oblique (a) and lateral oblique (b) 3D volume-rendered images show a double aortic arch with a dominant right arch and a small atretic portion (yellow arrow) in the left arch. There is tethering of the left subclavian artery (pink arrow) posteriorly opposite to an aortic dimple (black arrow) by the atretic segment (yellow arrow). Axial (c) and coronal (d) volume-rendered 3D images show a right dominant arch (green arrow). Note the compression on the airway (*) on the coronal (d) and virtual bronchoscopy (e) images. Most double aortic arches have a dominant right arch, and unlike this case, on coronal images it appears slightly more superiorly located. 3d volume rendered images make it easier to quickly evaluate arch dominance and location
DAA is rarely associated with congenital heart disease, although when present, is usually tetralogy of Fallot. When considering surgical correction, it should be kept in mind that either a ligamentum arteriosum or, less commonly, a patent ductus arteriosus, typically on the left side, may be present and should be divided in addition to one of the arches. If not, the ligamentum may still form a vascular ring once the arches are divided (Weinberg 2006; Weinberg and Whitehead 2010).
3.1.1.2 Right Aortic Arch Anomalies
There are three major types of right aortic arch anomalies associated with vascular rings. These anomalies include the following: (1) right aortic arch with an aberrant left subclavian artery off a Kommerell diverticulum (2) right aortic arch with left descending aorta (right circumflex aortic arch) and (3) right aortic arch with a mirror image branching and a left retroesophageal ductus arteriosus or ligamentum arteriosum.
The right aortic arch with an aberrant left subclavian artery off a Kommerell diverticulum is the second most common form of symptomatic vascular rings after double aortic arch (Figs. 4 and 5). In this anomaly, the right aortic arch gives rise to, in the order of occurrence, the left common carotid artery, the right common carotid artery, the right subclavian artery, and an aberrant left subclavian artery, which originates from a diverticulum of Kommerell. It is important to recognize that a diverticulum of Kommerell is associated with the presence of a left ligamentum arteriosum, which is not visible using current imaging modalities but that attaches the pulmonary artery to the aortic diverticulum constituting a vascular ring. The so-called diverticulum of Kommerell is the result of the embryonic origin of the left aberrant subclavian artery off the patent ductus arteriosus (Fig. 6). The ductus arteriosus carries a significant amount of flow during fetal life into the descending aorta, while the aberrant subclavian artery only carries a small amount of flow into the left upper extremity. Following birth and once the ductus involutes, the distal portion of the subclavian artery remains small, while the proximal portion, which originates from the ductus, remains relatively larger in size, resulting in this aortic diverticulum. Although the flow in the proximal and distal subclavian arteries is similar, the difference in caliber persists. Therefore, an aortic diverticulum implies the presence of a ligamentum arteriosum in the side of the diverticulum, and if located in the contralateral side of the aortic arch, a vascular ring can be inferred (Weinberg 2006; Weinberg et al. 1998; Weinberg and Whitehead 2010; Hellinger et al. 2011; Kondrachuk et al. 2012).
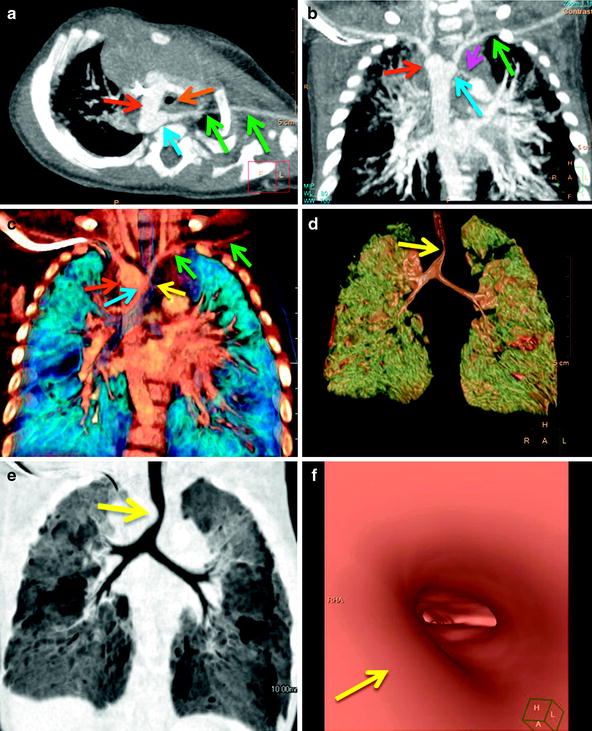
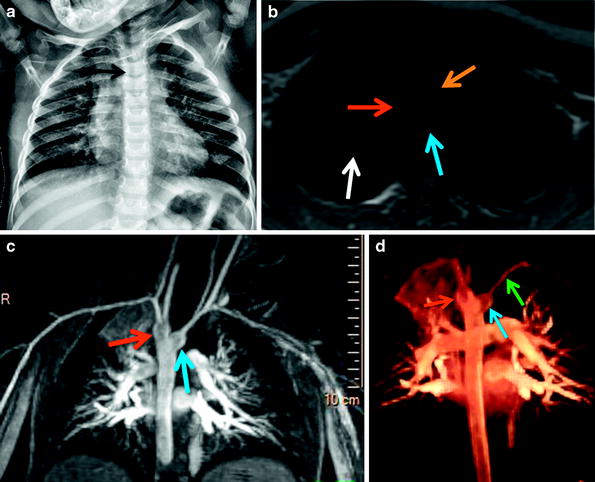

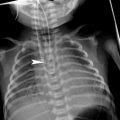
Fig. 4
A 5-month-old with noisy breathing since birth, who developed worsening stridor and significant respiratory distress with recurrent upper respiratory infections. Axial oblique (a) and coronal (b) maximum intensity projection and coronal volume-rendered (c) images show a right aortic arch (red arrows) with an aberrant left subclavian artery (green arrows) off a Kommerell diverticulum (blue arrows). Note the decrease caliber of the trachea (orange arrow) in (a) and the compression on the right wall of the trachea (yellow arrows) seen on the volume rendered (c, d), minimum intensity projection (e), and virtual bronchoscopic images. A tiny, patent left ductus arteriosus (pink arrow in b) is present confirming the presence of a vascular ring
Fig. 5
A 3-year-old with recurrent pulmonary infections underwent cardiac MRI for vascular ring evaluation noted on UGI (image not shown). (a) Frontal chest radiograph shows the presence of a right aortic arch (arrow) evidenced by mild effacement of the right wall of the trachea. (b) Axial dark-blood image shows a right aortic arch (red arrow) and the presence of a retroesophageal Kommerell diverticulum (blue arrow). Note the small caliber of the trachea (orange arrow) and the presence of right upper lobe airspace opacification (white arrow). Maximum intensity projection (c) and 3D volume-rendered (d) images depict a right aortic arch with an aberrant left subclavian artery (green arrows) arising from a prominent a diverticulum of Kommerell (blue arrow) and conforming a vascular ring. Note the difference in size between the diverticulum and the aberrant left subclavian artery
Fig. 6
Right aortic arch with left descending aorta in a 4-day-old presenting with respiratory distress. Axial oblique maximum intensity projection (a) and coronal volume-rendered (b) CT images shows the ascending aorta (AAo), a right-sided aortic arch (RAoA), and a retro esophageal distal circumflex aortic arch (yellow arrow) coursing into a left-sided descending aorta. Note the position of the descending aorta with respect to the spine. Posterior (c) and posterior oblique (d) 3D volume-rendered images demonstrate a prominent, patent ductus arteriosus (PDA) completing the ring. Note that an aberrant left subclavian artery (LSCA) originates from the PDA. Compare the caliber of the LSCA with that of the PDA, this explains the discrepant size of the LSCA with respect to any aortic, Kommerell diverticulum (asterisk), which is an embryologic remnant of the ductus. MPA main pulmonary artery; LPA left pulmonary artery; RPA right pulmonary artery; RSCA right subclavian artery; LSCA left subclavian artery
Conversely, in the case of a right aortic arch with an aberrant left subclavian artery, in which the caliber of the aberrant left subclavian vessel is uniform all along its course, no vascular ring can be anticipated. In these cases, the ductus or ligamentum arteriosum will presumably be right-sided, and it will not tether the aberrant left subclavian artery. Therefore, no complete, constrictive vascular ring is formed (Weinberg 2006; Weinberg and Whitehead 2010; Weinberg et al. 1998).
A right aortic arch with a left descending aorta (right circumflex aortic arch) and a left ductus or ligamentum arteriosum is the third most common type of vascular ring. In these cases, the arch courses behind the trachea and esophagus, so-called circumflex aortic arch, and then following an acute turn inferiorly the descending aorta courses along the left of the midline. This is unlike cases of right aortic arch in which the descending aorta after coursing over the right mainstem bronchus gradually descends for some distance on the right, and then progressively courses into the left before reaching the aortic hiatus (Weinberg and Whitehead 2010; Weinberg 2006; Weinberg et al. 2012).
A right aortic arch with mirror image branching and a left retroesophageal ductus arteriosus or ligamentum arteriosum is an uncommon anomaly, and the only type of right aortic with mirror image branching forming a vascular ring. The usual right aortic arch with mirror image branching, but without vascular ring, is typically seen in cases of congenital heart disease, usually tetralogy of Fallot. In right aortic arch with mirror image branching and a left retroesophageal ductus arteriosus or ligamentum arteriosum the branching pattern has the brachiocephalic artery (left common carotid and left subclavian arteries) as the first branch, followed by the right common carotid and the right subclavian arteries. The ring is completed by a patent ductus arteriosus or a ligamentum arteriosum originating typically from a prominent aortic diverticulum that courses leftward and behind the esophagus to reach the left pulmonary artery. As stated before, this anomaly may be easily confused with a right aortic arch with mirror image branching, which is typically found in patients with congenital heart disease, characteristically tetralogy of Fallot, but this anomaly does not constitute a vascular ring in the majority of the cases as in these instances, the ductus or ligamentum are usually right-sided. Less commonly, it may be left-sided, however, in these cases it most commonly originates from the base of the innominate artery and not from the aortic arch, consequently it does not encircle the airway and esophagus and does not form a vascular ring. If this is the case, one could expect to identify a quite prominent innominate artery, because of the large amount of blood flow being shunted from the left pulmonary artery and right heart across the patent ductus into the innominate artery during fetal life. Therefore, in the cases in which the ductus is closed, it is extremely helpful to identify the ductus dimple. If the ductus dimple is ipsilateral to the right aortic arch and directed toward the right, no ring is present, while if it is directed toward the left, the presence of a vascular ring can be inferred (Weinberg 2006; Weinberg and Whitehead 2010; Hellinger et al. 2011; Weinberg et al. 2012).
3.1.1.3 Left Aortic Arch Anomalies
A left aortic arch with an aberrant right subclavian artery is the most common arch anomaly, but it does not constitute a vascular ring, as the trachea and esophagus are not completely encircled by vessels and/or ligaments. In these cases, the arch gives rise to, in sequence, the right common carotid artery, the left common carotid artery, the left subclavian artery, and the right subclavian artery (arteria lusoria), which takes a retroesophageal course (Fig. 7). In older literature, the presence of this variant was reported to result in dysphagia in elderly patients. In these cases, the right aberrant subclavian artery is smooth in its contours, it is nearly equal in caliber throughout its intrathoracic course and tapers gradually (Weinberg 2006; Weinberg and Whitehead 2010). Very rarely, a left-sided aortic arch may coexist with a right Kommerell diverticulum giving origin to an aberrant right subclavian artery, indicating that a right-sided ligament is present, forming a complete vascular ring (Weinberg and Whitehead 2010; Kellenberger 2010).
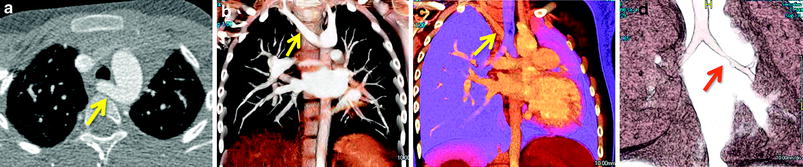
Fig. 7
History of recurrent left pneumonias and concern for vascular ring. (a) Axial CTA and (b, c) volume-rendered CT images reveal an aberrant right subclavian artery (yellow arrows), which is smooth in caliber and without evidence of a Kommerel diverticulum or airway compression. The presence or not of a Kommerell diverticulum and a vascular ring is typically better appreciated on 3D imaging. In this case there is no diverticulum, the aberrant subclavian vessel is smooth in contours and caliber and this type of arch anomaly does not constitutes a vascular ring. (d) However, volume-rendered CT image dedicated to the airway demonstrates a high-grade left mainstem bronchus narrowing (red arrow)
3.1.1.4 Pulmonary Artery Sling
In a pulmonary artery sling (PAS), the left pulmonary artery (LPA) arises from the posterior aspect of the right pulmonary artery, instead of the main pulmonary artery, and courses between the trachea and the esophagus to reach the left hilum, forming a sling around the distal trachea and the proximal right mainstem bronchus. The coexisting presence of a left ligamentum arteriosum connecting the main or right pulmonary artery and the left descending aorta results in a complete vascular ring that enwraps the trachea but spares the esophagus (Castañer et al. 2006; Hellinger et al. 2011; Lee et al. 2011a). It is believed that PAS is the result of proximal-left-sixth arch involution, with development of a secondary connection to the right-sixth branchial arch through the embryonic peritracheal primitive mesenchymal vessels (Newman and Ya 2010; Hellinger et al. 2011; Castañer et al. 2006).
There are two main types of PASs. In type I, the position of the carina is normally situated at the T4-5 level. In most instances, the airway is intrinsically normal with or without an associated tracheal bronchus. In these cases, the aberrant LPA potentially compresses the posterior wall of the distal trachea and the lateral aspect of the right mainstem bronchus, resulting in tracheobronchomalacia and leading to right lung air-trapping (Newman 2006; Newman and Ya 2010; Lee et al. 2011a). Type II is more common and is associated with a more inferiorly located carina at the T6 level; it is characteristically associated with long-segment tracheal stenosis with complete cartilaginous rings and abnormal bronchial branching, including a T-shaped carina and a right-bridging bronchus. Other cardiovascular, gastrointestinal, and right-lung anomalies may coexist, including lung hypoplasia, aplasia, agenesis, and scimitar syndrome (Lee et al. 2008a; Newman and Ya 2010). Patients with PAS characteristically present as infants with respiratory symptoms, such as stridor, apneic spells, or hypoxia. The timing and severity of the symptoms are dictated by the severity of the accompanying airway abnormalities, which may be triggered by an acute upper respiratory infection (Newman and Ya 2010; Newman 2006).
Imaging findings in PASs depend on its type and other coexisting congenital anomalies. In type I, significant right-sided hyperinflation due to partial obstruction and right bronchomalacia may be appreciated. Although the right lung may be fluid-filled and appear radio-dense in the early neonatal period due to prolonged fetal fluid retention, a right-sided tracheal bronchus above the carina may occasionally be observed. On occasion, on the lateral projection in both types, a small, rounded, soft-tissue density may be present between the mid trachea and esophagus, representing the LPA coursing between these two structures. In type II PASs, bilateral hyperinflation may be observed in cases of long-segment tracheal stenosis. In cases of unexplained right-sided volume loss, if the trachea appears narrow or difficult to see and the carina appears low and horizontal on frontal chest radiographs, these findings should raise suspicion of type II PAS.
MDCT and a cardiac MRI with 3D reconstructions are excellent imaging tests for the assessment of PASs, because the origin, size, and entire course of the aberrant LPA, as well as the associated central airway anomalies, can be accurately depicted on the 3D renderings on both modalities (Figs. 8 and 9). When concomitant lung anomalies are suspected, a CT should be obtained for accurate assessment of the lung parenchyma; the MRI resolution is still not useful, although the gap is narrowing. Paired inspiratory/expiratory CT scans can accurately demonstrate tracheobronchomalacia often associated with PAS (Hellinger et al. 2011; Lee et al. 2008b).
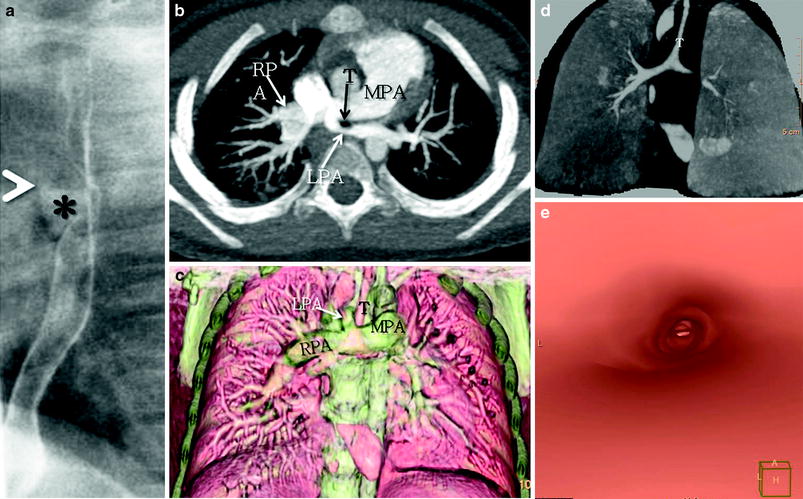
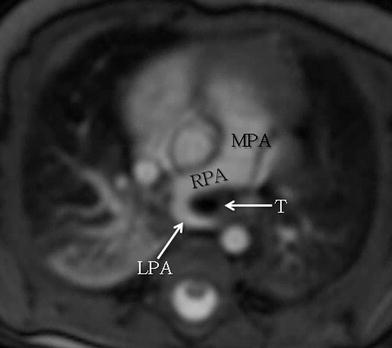
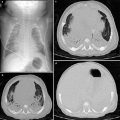
Fig. 8
A 3-year-old female with a history of trisomy 21 and stridor. a. Lateral esophagogram image shows a classic anterior indentation (*) on the esophagus with narrowing and anterior displacement of the distal trachea (T). Axial maximum intensity projection (b) and coronal volume rendered (c) CT images show anomalous origin of the left pulmonary artery (LPA) from the right pulmonary artery (RPA) instead of the main pulmonary artery (MPA) and coursing between the trachea and esophagus forming a sling around the trachea. Coronal minimum intensity projection (d) and virtual bronchoscopy (e) CT images show findings consistent with long-segment tracheal stenosis and a rounded appearance of complete tracheal rings. There is in addition complete effacement of the left mainstem bronchus, the result of a combination of vascular compression as the bronchus courses between the left main pulmonary artery and descending aorta, and tracheobronchomalacia
Fig. 9
Axial white-blood MRI image shows anomalous origin of the left pulmonary artery (LPA) off the right pulmonary artery (RPA) instead from the main pulmonary artery (MPA) with imaging findings consistent with LPA sling
Asymptomatic patients may be followed clinically. Patients with type I PAS and respiratory symptoms may benefit from re-implantation of the LPA and excision of the patent ductus arteriosus or ductal ligament. In type II PAS, re-implantation or anterior translocation of the LPA alone will not result in improvement of respiratory symptoms if the long-segment airway stenosis is not addressed, usually by slide tracheoplasty. In mildly symptomatic cases, there are some anecdotic reports of spontaneous improvement (Newman 2006; Newman and Ya 2010).
3.1.2 Obstructive Lesions of the Aortic Arch
The aortic arch is divided into three main segments: (1) the proximal transverse arch, extending from the origin of the innominate artery to the left common carotid artery; (2) the distal transverse arch, extending from the left common carotid artery to the left subclavian artery; and (3) the aortic isthmus, which is the aortic segment extending distal from the left subclavian artery to the ligamentum or ductus insertion (Backer and Mavroudis 2000; Restrepo et al. 2012). In general, all three major supraaortic branches, the brachiocephalic artery, the left common carotid artery and the left subclavian artery originate off the aortic arch fairly close together, and a distance between each segment exceeding the 5 mm is considered abnormal (Moulaert et al. 1976).
Obstructive abnormalities may affect the aortic arch or its segments. The obstruction may be in the form of a discrete, shelf-like coarctation, typically in a juxtaductal location and affecting a focal area of the arch versus a diffuse, smooth, tubular hypoplastic narrowing involving a longer portion of the arch (Matsui et al. 2007). The narrowing becomes significant when a pressure gradient is measured across the area of narrowing, and it usually occurs when there is more than a 50 % reduction in luminal cross-sectional area. Although gradual tapering of the aortic isthmus is normal up to 3 months after birth, persistence beyond this limit should be considered abnormal (Ho and Anderson 1979; Restrepo et al. 2012).
There are four main types of obstructive lesions of the aortic arch (Matsui et al. 2007; Epelman and MacDonald 2010): (1) Discrete coarctation of the aorta, (2) Tubular hypoplasia of the aortic arch, (3) Combined hypoplasia and discrete coarctation, which usually coexist in approximately 30 % of instances (Brown et al. 2009), and (4) Interruption of the aortic arch (Matsui et al. 2007).
3.1.2.1 Coarctation of the Aorta
Coarctation of the aorta is the 6th most common type of congenital heart disease, accounting for approximately 7 % of all congenital heart anomalies in children with a male–female ratio of 2:1 (Tanous et al. 2009; Restrepo et al. 2012; Murillo et al. 2012). Coarctation of the aorta is defined as a hemodynamically significant, focal, shelf-like narrowing of the descending aorta typically distal to the takeoff of the left subclavian artery in the region of the ligamentum arteriosum. The shelf of the coarctation is most often juxtaductal, but it may be pre- or postductal (Backer and Mavroudis 2000; Tanous et al. 2009; Murillo et al. 2012). Coarctation of the aorta may be a feature of Turner’s syndrome and is associated with a bicuspid aortic valve in more than 70 % of patients (Tanous et al. 2009; Murillo et al. 2012). Other intracardiac abnormalities may also be found in association with coarctation, especially in patients who present in infancy and are more likely to have associated left ventricular outflow obstruction and/or associated ventricular septal defects, which may be perimembranous, muscular or misalignment. Mitral valve abnormalities resulting in mitral stenosis, such as a supravalvular mitral ring, dysplastic mitral valve leaflets, or a parachute mitral valve may also be present in patients with coarctation. Subaortic stenosis is also a potentially associated lesion. When this group of left-sided obstructive lesions occurs together, they are referred to as the Shone complex (Moene et al. 1987; Rosenquist 1974; Shone et al. 1963; Moulaert et al. 1976).
Unless corrected in infancy, collateral circulation gradually develops between the proximal aorta and the distal aorta to bypass the coarctation. The vessels contributing to the collateral circulation may become markedly enlarged and tortuous by early adulthood (Hellinger et al. 2011; Restrepo et al. 2012).
The age of presentation and symptoms depends on the severity, rather than on the site, of the coarctation, the presence or absence of associated lesions and, most importantly, the patency of the ductus arteriosus, as the ductus acts as a bypass for blood distal to the coarctation. Once the ductus closes, in cases of high-grade stenosis, congestive heart failure symptoms develop. The blood pressure is elevated in the vessels originating proximal to the coarctation; the blood pressure as well as the pulse pressure below the coarctation are typically lower (Epelman and MacDonald 2010; Jenkins and Ward 1999). Coarctation of the aorta recognized after infancy is rarely associated with significant symptoms. Older children are frequently referred to a cardiologist when they are found to be hypertensive on routine physical examination. Arterial pulses in the legs are either absent or weak and delayed, and there is hypertension in the arm, or blood pressure readings in the arm are higher than those in the leg. In normal children, the systolic pressures in the thigh or calf are higher by 5–10 mm Hg than that in the arm.
If aortic coarctation is left untreated, it is expected that 90 % of the patients will die before the age of 50 (Campbell 1970). The most common causes of death are congestive heart failure, aortic rupture, bacterial endocarditis, and intracranial hemorrhage (Campbell 1970).
On chest radiography, the findings will vary according to the severity of the coarctation and the presence of the associated pathology. Abnormality in the contour of the isthmus region is the most important sign in coarctation, and it may be the only finding. The contour of the proximal descending aorta may resemble a number 3 (“Figure of 3 sign”); the upper portion of the 3 is related to the combined shadows of the pre-stenotic aortic knob and the dilated proximal portion of the left subclavian artery; the waist of the “figure of 3” represents the coarctation itself; and the lower portion corresponds to the poststenotic dilatation of the descending aorta (Epelman and MacDonald 2010). Rib notching between the fourth and eighth ribs may be observed in older children but rarely before the age of seven. Rib notching is the result of pressure erosion caused by dilated or tortuous intercostal arteries, which serve as collaterals between the internal mammary arteries and the descending aorta. The first, second, and third ribs do not typically exhibit rib notching, as the intercostal arteries at these level originate from the thyrocervical trunk, which originates above the coarctation (Epelman and MacDonald 2010).
Critical preoperative information to be evaluated with cross-sectional imaging (Figs. 10 and 11) includes (1) the anatomy of the transverse aorta and the aortic isthmus, including the orthogonal dimensions and the relationship to other arch vessels; (2) the presence and extent of any pre-coarctation hypoplasia; (3) the severity of the obstruction and hemodynamics, including quantification of collateral flow, flow velocity, flow volume, and pressure gradients across the stenosis, by phase contrast MR imaging; (4) the presence of previously mentioned associated intracardiac anomalies, such as bicuspid aortic valve and mitral valve abnormalities (Konen et al. 2004); and (5) the presence of aneurysm, particularly in cases after prior surgical or endovascular interventions (Weinberg and Whitehead 2010; Restrepo et al. 2012; Konen et al. 2004; Hom et al. 2008).
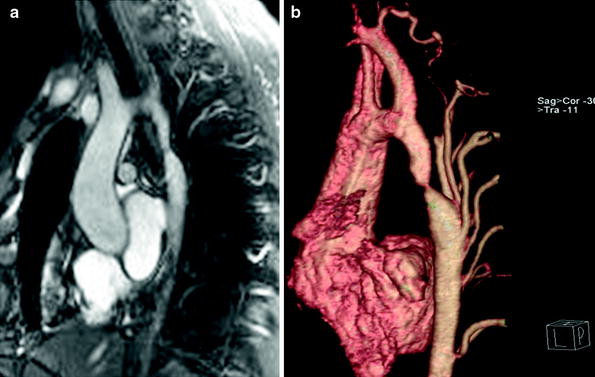
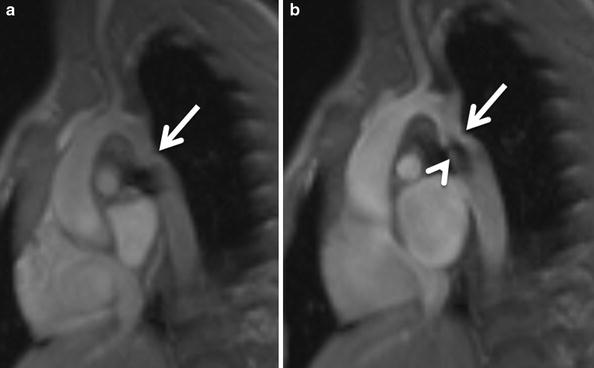
Fig. 10
Sagittal oblique (candy-cane) white-blood (a) and volume-rendered (b) MR images show a discrete aortic coarctation (arrow) with multiple hypertrophied collaterals
Fig. 11
A 4-year-old with a moderate discrete coarctation of the aorta. Diastolic (a) and systolic (b) frames of steady-statefree precession MR images show a posterior indentation at the coarctation site (arrow). Note signal void (arrowhead) due to turbulent flow at the coarctation site
While surgery is the mainstay of treatment in infants, endovascular repair with balloon angioplasty and stenting is being increasingly used, particularly in older children (Holzer et al. 2010). Commonly used surgical procedures include resection and end-to-end anastomosis, subclavian flap aortoplasty, synthetic patch repair, and aortic bypass interposition grafting or bypass grafting (Jenkins and Ward 1999). The continued monitoring of patients with stents is required to assess for complications such as aneurysm formation or the need for reintervention (Holzer et al. 2010).
3.1.2.2 Tubular Hypoplasia of the Aortic Arch
Tubular hypoplasia of the aortic arch may involve one or all segments and characteristically presents in association with other intracardiac anomalies, ranging in severity from a bicuspid aortic valve to a more complex left-sided obstructive lesion or hypoplastic left heart syndrome (Brown et al. 2009). A common rule of thumb for determining aortic arch hypoplasia in neonates on echocardiography is an internal luminal diameter of less 1 mm/kg (Brown et al. 2009). This finding may coexist with associated discrete, focal coarctation in nearly 30 % of the cases (Brown et al. 2009). The narrowing is smooth and usually affects the isthmus, although in rare occasions, it may affect the entire arch (Brown et al. 2009; Matsui et al. 2007). Recognition of this instance is important, as it may affect the operative approach, and in some complex patients, the surgeon may opt for a median sternotomy instead of the classic left thoracotomy (Brown et al. 2009) (Fig. 12).
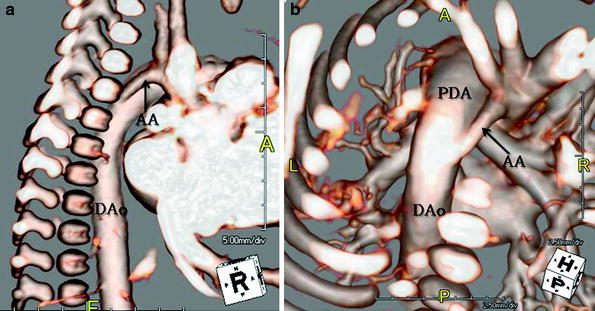
Fig. 12
A 4-day-old with tubular hypoplasia of the aortic arch (AA). Lateral (a) and (b) cranial posterior volume rendered MR images show diffuse, severe narrowing of the transverse aorta and isthmus proximal to the patent ductus arteriosus (PDA). The ductus is quite prominent in size and is similar in caliber to the descending aorta (DAo) as the systemic circulation is ductal dependent
3.1.2.3 Interruption of the Aortic Arch
Interruption of the aortic arch (IAA) is an uncommon, hemodynamically critical, obstructive aortic arch anomaly. In this condition, the left and right fourth embryonic arches have regressed and there is lack of luminal continuity between the ascending and descending aorta. It accounts for 1.5 % of all congenital heart disease. In IAA, the flow between the ascending and descending aorta and the lower body is maintained only by a ductal arch (patent ductus arteriosus) or collateral circulation once the ductus closes (Kellenberger 2010; Hellinger et al. 2011; Weinberg and Whitehead 2010; Weinberg et al. 1998; Mishra 2009). The affected neonates are typically not cyanotic and normal appearing at birth, but they present usually later on, characteristically during the first 2 weeks of life, with congestive heart failure that worsens as the ductus closes (Hellinger et al. 2011; Kellenberger 2010).
The classification system proposed by Celoria and Patton (Celoria and Patton 1959) is the most widely accepted and classifies these anomalies in three types according to the point of interruption: (1) in type A, the interruption occurs distal to the left subclavian artery origin at the aortic isthmus; (2) in type B, the interruption takes place between the origins of the left common carotid artery and the left subclavian artery (Fig. 13); and (3) in type C, the interruption takes place proximal to the left common carotid artery, between the origins of the innominate and left common carotid arteries (Weinberg and Whitehead 2010; Kellenberger 2010; Hellinger et al. 2011). Type B is the most common type, and it accounts for approximately two-thirds of the cases. Type C is the least common type, occurring in less than 2 % of the cases (Collins-Nakai et al. 1976; Norwood et al. 1983; Vouhe et al. 1990; Jonas et al. 1994; Serraf et al. 1996; Tlaskal et al. 1998; Brown et al. 2006; Flint et al. 2010; Hellinger et al. 2011). Similar to other arch anomalies involving the fourth embryonic arch, Type B interruption is commonly observed in patients with DiGeorge syndrome (Goldmuntz et al. 1998; Momma 2010). In addition, IAA rarely occurs in isolation and a patent ductus arteriosus is always present. VSD, as well as a left ventricular outflow tract obstruction, are commonly present. Less commonly, IAA may be associated with aorto-pulmonary window, truncus arteriosus, and double outlet right ventricle (Mishra 2009; Restrepo et al. 2012). These patients are surgically treated early in life; if left untreated, death may ensue at a median age of 4–10 days (Kellenberger 2010; Mishra 2009). Imaging should be tailored to identify the precise type of IAA, to measure the distance between the segments, to assess the dimensions of the left ventricular outflow tract, and to describe the morphological characteristics of the patent ductus arteriosus and any associated anomalies.
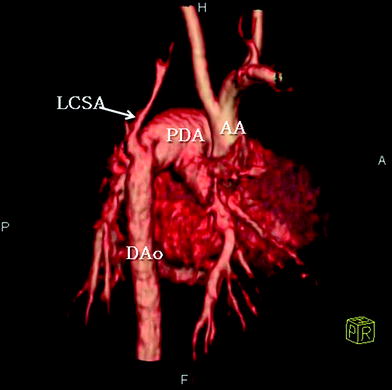
Fig. 13
A 11-day-old patient with interrupted aortic arch. Volume-rendered CT image shows complete lack of continuity between the proximal aortic arch (AA) and the descending aorta (DAo), with imaging findings consistent with aortic arch interruption between the origins of the left common carotid artery and the left subclavian artery (LSCA). The descending aorta is reconstituted via a patent ductus arteriosus (PDA)
3.1.3 Shunt Lesions
3.1.3.1 Patent Ductus Arteriosus
Patent ductus arteriosus (PDA) is the most common heart defect in neonates, and it is defined as incomplete closure of the ductus arteriosus beyond functional closure after birth. Its incidence is quite high in premature infants, being as high as 60 % in those born before 28 weeks of gestation (Corno and Festa 2009b). PDA consists of a vascular connection usually between the left proximal pulmonary artery and the descending thoracic aorta (Fig. 14), just distal and opposite to the left subclavian artery ostium bypassing the deflated lungs. However, the precise point of connections can vary substantially (Backer and Mavroudis 2000; Berko and Haramati 2012; Kondrachuk et al. 2012). Rarely, the PDA can insert onto the right pulmonary artery. The PDA usually connects to the left pulmonary artery, even in the presence of a right aortic arch. Bilateral patent ducti have also been rarely described (Corno and Festa 2009b). The ductus arteriosus usually closes within 72 h of birth, but if it does not, it is considered unlikely that it will close spontaneously (Berko and Haramati 2012). It typically closes beginning from the pulmonary end, leaving a diverticulum on the aortic side, which eventually closes as well (Corno and Festa 2009b). This anomaly usually persists in patients with underlying congenital heart disease, commonly ASD or VSD, but it is also seen in diseases that are ductus-dependent, such as hypoplastic left heart syndrome, aortic arch interruption, and pulmonary atresia with or without an intact ventricular septum (Corno and Festa 2009b). On cross-sectional imaging, the ductus arteriosus appears as a tubular structure joining the aorta and the proximal left pulmonary artery (Fig. 14), which can be easily missed if only axial imaging is used (Kondrachuk et al. 2012). Conversely, the location, caliber, and morphology of the PDA can be easily depicted with multiplanar reconstructions in isolated defects or in complex cardiac malformations. PDA in older patients may be potentially complicated by aneurysm formation, calcification, and rupture. An increased risk for bacterial endarteritis has also been reported (Moore and Brook 2012; Berko and Haramati 2012). Premature infants are given a trial of prostaglandin inhibitors. If this fails, surgical ligation is performed via a left posterolateral thoracotomy. Percutaneous coil occlusion is reserved for older children (Kondrachuk et al. 2012; Corno and Festa 2009b).
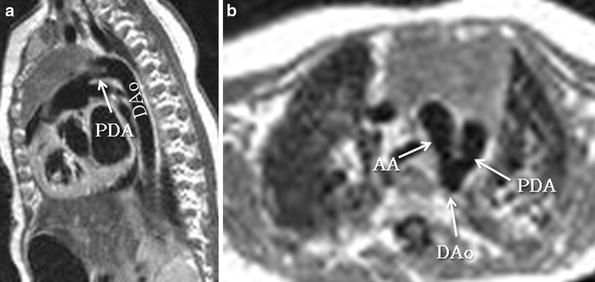
Fig. 14
Patent ductus arteriosus in a 3-month-old girl. Sagittal oblique (candy cane) and axial black-blood MR images show a patent ductus arteriosus (PDA) connecting the pulmonary artery with the descending aorta (Dao). AA Aortic arch
3.1.3.2 Aortopulmonary Collaterals
Major aortopulmonary (systemic to pulmonary) collateral arteries (MAPCAs) characteristically develop in the setting of severe pulmonary stenosis/atresia or right ventricular outflow tract obstruction serving as the primary source of blood flow to the lungs (Kondrachuk et al. 2012; Flamm 2010). MAPCAs are highly variable in their presence, size, and origin. They may originate more frequently from the descending aorta or, less frequently, from the arch vessels, or the subclavian, internal mammary or intercostal arteries (Lofland 2004; Kondrachuk et al. 2012). MAPCAs may be diminutive or extremely large, resulting in over-circulation to the portion of the lung supplied by them. They may exhibit focal areas of stenosis either at their origin or anywhere along their course. MAPCAs may also have connections with the native pulmonary arteries (Lofland 2004, 2009). Multidetector CT or contrast-enhanced MRI with 3D reconstructions are extremely helpful for their full depiction and mapping prior to unifocalization or interventional embolization (Fig. 15) (Corno and Festa 2009c; Lofland 2004, 2009).
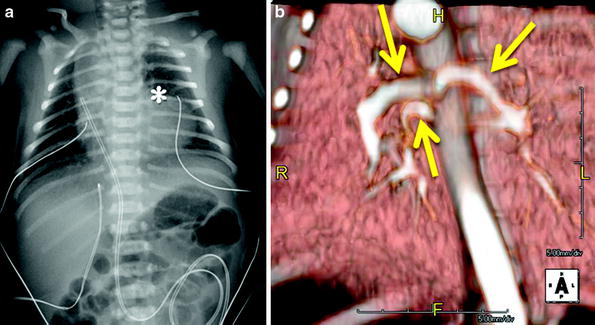
Fig. 15




Newborn with respiratory distress. (a) Frontal chest radiograph shows moderate cardiomegaly on account of the right chambers, an absent main pulmonary artery segment (asterisk), and decreased peripheral pulmonary vascularity. Echocardiography demonstrated tetralogy of Fallot. (b). Volume-rendered CTA image was obtained to map out the aortopulmonary collateral arteries (arrows) prior to unifocalization and demonstrates three prominent MAPCAs supplying both lungs
Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
