Primary Pulmonary Tumors
Benign
Malignant
Hamartoma
Inflammatory myofibroblastic tumor
Chondroma
Pleuropulmonary blastoma
Fetal lung interstitial tumor
Bronchial carcinoid
Infantile myofibromatosis
Mucoepidermoid carcinoma
Congenital peribronchial myofibroblastic tumor
NUT midline carcinoma
Papilloma
Bronchogenic carcinoma
Leiomyoma
Leiomyosarcoma
4.1 Pulmonary Metastases
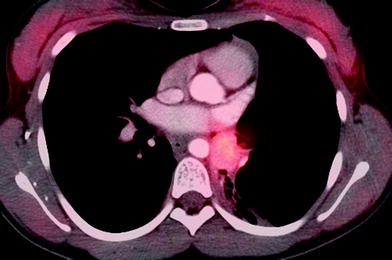
The lungs are a common site of metastatic spread of many pediatric solid extracranial malignancies, including (with an approximate percentage incidence of pulmonary metastases at diagnosis) Wilms tumor (10 %), osteosarcoma (15–20 %), Ewing sarcoma (15–20 %), rhabdomyosarcoma (15 %), and hepatoblastoma (10 %) (Paulussen et al. 1998; Kaste et al. 1999). Pulmonary metastases are found in only 1–3 % of cases of neuroblastoma at diagnosis (Cowie et al. 1997), and in 3–4 % of those with stage 4 disease (DuBois et al. 2008), but are a prognostic marker of unfavorable outcome (Kammen et al. 2001; DuBois et al. 2008), as is the case for nearly all pediatric solid extracranial malignancies (Fuchs et al. 2012). Routine chest CT for staging purposes is warranted at diagnosis for these malignancies, as well as for some other less common pediatric malignancies. Staging for metastatic neuroblastoma is, however, best achieved with metaiodobenzylguanidine (MIBG) scanning. Lung metastases usually result from hematogenous dissemination and manifest as multiple, well-defined pulmonary nodules. Lymphatic spread producing a reticular or reticulonodular pattern can also occur in neuroblastoma (McCahon 2006) (Fig. 1).
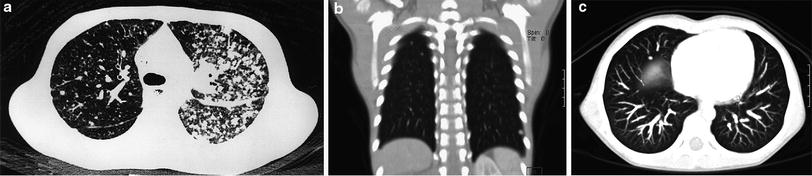
Fig. 1
Pulmonary metastases. a Axial chest CT image demonstrates predominantly left-sided pulmonary nodularity, pleural and interstitial thickening due to neuroblastoma metastases. An unusual lymphatic pattern of tumor spread has occurred in this patient. b Two subtle right apical pulmonary nodules and another nodule in the left lower lobe depicted on a coronal CT image are suspicious for metastatic disease in a different 4-year-old patient with a newly diagnosed renal tumor. c Axial MIP CT image from the same patient illustrating how the MIP technique clearly helps differentiate between pulmonary vessels and small lung nodules
Diagnostic dilemmas arise when pulmonary nodules are small (a few millimeters in diameter), indistinct, or solitary. Among malignant causes, an incidentally detected nodule is much more likely to represent a metastasis rather than a primary lung tumor in a child. Benign causes of pulmonary nodules are myriad and include granulomas (particularly in areas with a high prevalence of histoplasmosis or tuberculosis), hamartomas, chondromas, intraparenchymal lymph nodes, septic emboli, plugged distal airways, previous varicella infection, vasculitis, Langerhans cell histiocytosis, respiratory papillomatosis, opportunistic infections in the setting of immunodeficiency, and fibrotic nodules in the setting of bleomycin or cyclophosphamide toxicity. Clustering of multiple nodules favors an infectious or inflammatory etiology. Spontaneous resolution of pulmonary nodules may be related to a focal inflammatory process, atelectasis, mucoid impaction of small bronchi, or intermittent enlargement of benign intrapulmonary lymph nodes (Diederich et al. 2005). Intrapulmonary lymph nodes tend to be located peripherally below the level of the carina at or near the junction of the pleura and adjacent lung lobules (Wang et al. 2013) (Fig. 2).
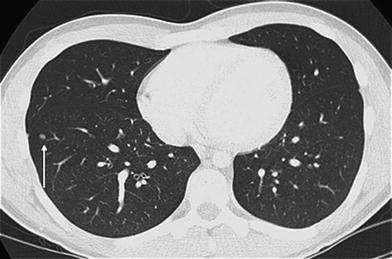
Fig. 2
Intrapulmonary lymph node. Axial chest CT image from a 17-year old with osteosarcoma undergoing staging demonstrates a well-circumscribed, bean-shaped, solid pulmonary nodule (arrow) of the right lower lobe close to the pleura. This nodule proved to be an intrapulmonary lymph node rather than a metastasis upon biopsy
In an individual case, it can be very difficult to distinguish benign from metastatic pulmonary nodules in young patients with a solid extracranial malignancy. A study of pediatric oncology patients found that the most significant predictors of a pulmonary nodule being metastatic were peripheral location, size between 5 and 10 mm diameter, location in the right lower lobe, and a history of osteosarcoma, Ewing sarcoma, or hepatocellular carcinoma (Murrell et al. 2011). Another study found that sharp nodule margins and development of new nodules are significantly associated with malignancy, but that nodules measuring less than 5 mm in diameter are as likely to be malignant as larger nodules, unlike in adults (McCarville et al. 2006). Co-existence of both benign and malignant nodules is also possible. In the setting of pediatric osteosarcoma, over 90 % of malignant nodules are correctly classified, while only 11–30 % of benign nodules are correctly classified on the basis of imaging findings (Brader et al. 2011).
FDG-PET may be a useful adjunct for the evaluation of possible pulmonary metastases in pediatric malignancies. FDG-PET confers significantly higher specificity than CT for pulmonary metastases greater than 5 mm diameter in children (Kleis et al. 2009). A prospective study of FDG-PET/CT in the characterization of pulmonary nodules in children with bone sarcoma demonstrated a sensitivity of 90 % and specificity of 94 % using a SUVmax value around 1, compared to a sensitivity of 90 % and specificity of 81 % using a cut-off value of 6 mm for nodule diameter (Cistaro et al. 2012).
Findings equivocal for pulmonary metastatic disease on chest CT warrant further evaluation, either with FDG-PET, close surveillance with follow-up imaging, or biopsy. Because of the survival advantage conferred by complete resection of pulmonary metastases in selected pediatric malignancies (Tronc et al. 2008) and the difficulty in reliably diagnosing metastatic disease solely by imaging when only one or a few small lung nodules are discovered in a child with a solid tumor, there is a trend toward resecting or performing a biopsy of indeterminate pulmonary nodules. Fine needle aspiration cytology is widely practiced in adult patients with carcinoma and is usually diagnostic. In children, however, sarcomas are much more common, and aspiration cytology is generally regarded as unreliable for the diagnosis of childhood tumors since differentiation from other cells can be extremely difficult on small cytological specimens and architectural information is lost. Core needle biopsies of small pulmonary nodules in children under CT guidance using a co-axial system permit multiple biopsies through a single pleural pass to be obtained, yielding adequate tissue for diagnosis in most cases and reducing the likelihood of complicating hemothorax or pneumothorax (Connolly et al. 1999).
A successful response to chemotherapy should be accompanied by disappearance of pulmonary metastases. Occasionally, nodules may demonstrate incomplete resolution related to residual fibrosis or selection of less mitotically active cells (as in the case of rhabdomyomatous differentiation in Wilms tumors or mature teratoma elements in germ cell tumors), but ultimately proof of a benign nature rests on either biopsy or stability on follow-up, particularly off treatment (Seemayer et al. 1997; Seifert et al. 2012). If the initial chest CT at diagnosis is negative for metastases, later follow-up for pediatric oncology patients is largely with CXR, with CT reserved for equivocal CXR findings, clinically suspected relapses, or tumors with a high risk of pulmonary relapse.
Patients with osteosarcoma or FDG-PET positive disease at initial presentation appear to be at the highest risk of recurrent pulmonary metastatic disease (Murrell and Dasgupta 2013). As up to 30–40 % of children with osteosarcoma eventually develop pulmonary metastases and complete surgical remission is the main prognostic factor affecting survival, routine chest CT surveillance, and pulmonary metastasectomy of osteosarcoma patients may be justified (Diemel et al. 2009). In an osteosarcoma patient, pulmonary nodules, particularly those that are calcified or greater than 5 mm in diameter, should be regarded as malignant until proven otherwise (McCahon 2006; Brader et al. 2011). CT also tends to underestimate the number of metastatic pulmonary nodules found at thoracotomy in osteosarcoma patients (Kayton et al. 2006). Centrally-located pulmonary metastases of osteosarcoma may be associated with reduced median survival compared with those that are peripherally sited (Letourneau et al. 2011). Osteosarcoma may also metastasize to the mediastinal lymph nodes and pleura, and even to the myocardium. Cavitary metastases are unusual in childhood but are occasionally seen with sarcomas or Wilms tumor, or after chemotherapy or radiotherapy.
Co-operative pediatric oncology groups in North America and Europe once recommended that presumed pulmonary metastases from Wilms tumor could be ignored if they were not visible on CXR. This is no longer the case with current protocols from the Children’s Oncology Group (COG) and the International Society of Pediatric Oncology (SIOP). However, the clinical significance of these small nodules that are detectable only with CT in patients with Wilms tumor is uncertain. A retrospective review of National Wilms Tumor Studies (NWTS)-4 and -5 found that patients with pulmonary nodules detected only by CT who received three chemotherapeutic agents had improved 5 year event-free survival, but not overall survival, compared with those receiving only two drugs (Grundy et al. 2012). These so-called CT-only patients represent a small (2–4 %) cohort of all Wilms patients (Smets et al. 2012). It has been argued that this small proportion of CT-only detected lesions makes it difficult to justify the routine use of a relatively high radiation technique such as CT when patient outcomes may not be affected by the CT findings in the setting of Wilms tumor (Smets et al. 2012). In a SIOP Wilms study, patients with CT-only lesions should have been treated as having localized disease but many were not, leading the authors to conclude that current clinical decision-making was based on a fear of under-treatment. The outcome for patients with CT-only lesions, whether treated as localized or metastatic disease (the latter received more intensive therapy), were no different. Some patients with CT-only lesions may have benign chest disease, yet suffer late toxic effects from more intensive therapy. This underscores the need to devise better diagnostic imaging tests for the detection of metastases and selection of patients for more intensive therapy. Chest CT perhaps could be used in children with Wilms tumor after preoperative chemotherapy and nephrectomy to select those with persisting pulmonary lesions. The finding of persisting pulmonary lesions in combination with high risk histology and/or stage III disease confers a worse prognosis, and therefore these patients might benefit from intensified therapy (Smets et al. 2012). Wilms tumor patients with metastatic lung disease at presentation who are in complete pulmonary remission after chemotherapy with or without metastasectomy have a better outcome than those with unresectable pulmonary metastases. The findings on chest CT help determine which patients should or should not receive radiotherapy, with persisting lung lesions being an indication for radiotherapy (Verschuur et al. 2012).
An aggressive diagnostic approach with chest CT is also advocated in pediatric malignant non-Wilms renal tumors, where survival requires complete clearance of lung metastases, either through chemotherapy, radiotherapy, or metastasectomy (Warmann et al. 2012).
4.2 Pulmonary Lymphoma and Post-transplantation Lymphoproliferative Disorder
Pulmonary involvement occurs in approximately 10 % of cases of pediatric lymphoma and is usually associated with ipsilateral hilar or mediastinal lymphadenopathy. Pulmonary lymphoma is most frequently seen in Hodgkin disease or anaplastic large cell non-Hodgkin lymphoma, and most often manifests as nodules or consolidative masses. A reticular pattern related to lymphatic spread can also be observed (Maturen et al. 2004). In the setting of pediatric Hodgkin lymphoma, direct contiguous involvement of the lung by an adjacent mediastinal mass is considered stage IIE invasive disease rather than disseminated stage IV disease (Guillerman et al. 2011).
Post-transplantation lymphoproliferative disorder (PTLD) is thought to result from an Epstein–Barr virus (EBV)-induced proliferation from B-cell lymphocytes, which is normally opposed by a functioning T-cell system in immunocompetent patients. It is estimated to occur in 2–3 % of all solid organ transplants including both children and adults, but the incidence in children with lung transplants is reported to be 8 % (Pickhardt et al. 2000). Post-transplantation lymphoproliferative disorder (PTLD) occurs more often and presents earlier in lung transplants compared with other solid organ transplants, with a median time from transplant to diagnosis of 6–10 months (Wilde et al. 2005; Siegel et al. 2003). The overall prevalence of pulmonary involvement in pediatric PTLD is 29 % (Maturen et al. 2004), and a large majority of pediatric lung transplant recipients with PTLD have intrathoracic involvement of the lungs or mediastinum (Wilde et al. 2005).
The most common presentation of intrathoracic PTLD is asymptomatic pulmonary nodules. Close monitoring of EBV viral load and the degree of immunosuppression via lymphocyte function assays can facilitate an early diagnosis (Elidemir et al. 2009). Children at greatest risk are those who receive EBV-positive donor lungs who were EBV-seronegative prior to transplantation. The typical CT appearance is multiple well-defined, soft-tissue attenuation pulmonary nodules measuring 1–4 cm. They are usually homogeneous but can have central necrosis. In approximately 10 % of cases, multifocal masses or consolidations are the predominant parenchymal finding. Mediastinal lymphadenopathy is often present, and may coalesce into large nodal masses (Wilde et al. 2005).
The principal initial treatment of PTLD is cessation or reduction of immunosuppression. However, PTLD may progress to an aggressive diffuse large B-cell lymphoma (Fig. 3). Only 50 % of pediatric lung transplant patients were reported to survive more than 2 years after diagnosis of PTLD in one study (Siegel et al. 2003), but that figure has likely improved in recent years.
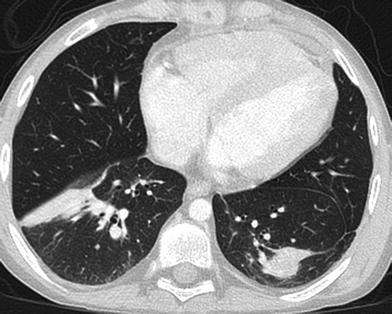
Fig. 3
Post-transplantation lymphoproliferative disorder (PTLD). Axial chest CT image from a 15-year-old post lung transplantation demonstrates lobular subsegmental masses of the lower lobes. Biopsy revealed monomorphic PTLD, diffuse large B-cell lymphoma type
4.3 Pulmonary Hamartoma
Hamartomas account for the majority of benign tumors that occur in the lung parenchyma in childhood (Hancock et al. 1993). A pulmonary hamartoma consists of variable mesenchymal tissues that are native to the lung but present in an abnormal configuration. Despite being regarded as possible developmental anomalies, the vast majority of pulmonary hamartomas are discovered in adults and grow very slowly (doubling time longer than 550 days in almost all cases) (Huang et al. 2011), supporting the idea that hamartomas are actually acquired lesions.
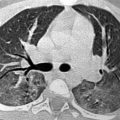
Pulmonary hamartomas in adults are frequently asymptomatic and often found incidentally (Hartman and Shochat 1983). The characteristic radiological finding is a clearly defined solid nodular opacity in the lung periphery. Hamartomas measuring less than 1 cm in diameter are more likely to be spherical, and those measuring over 1 cm in diameter are more likely to be lobulated. Approximately 10 % show calcification, often with a speckled or curvilinear configuration (Fig. 4), and sometimes in a “popcorn” pattern. Central fat attenuation is seen in up to 30 % of hamartomas on CT. The likelihood of identifying fat or calcification increases with lesion size and these classic features are rarely seen in lesions measuring less than 2 cm in diameter. Hamartomas typically show no uptake on FDG-PET (Huang et al. 2011). MRI reveals linear or curvilinear clefts along the marginal surface, particularly on T2-weighted imaging. Most lesions demonstrate cleft and rim enhancement with contrast, but fat is usually inconspicuous on MRI (Park et al. 2008).
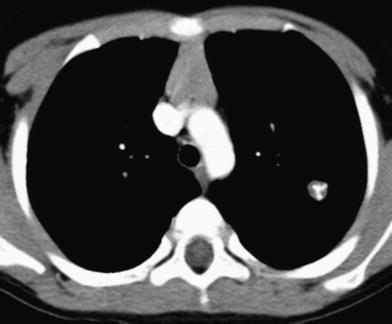
Fig. 4
Pulmonary hamartoma. Axial contrast-enhanced chest CT image shows a small, well-defined, lobular, left pulmonary nodule with curvilinear calcification
In contrast to adult patients, many of the reported cases of pulmonary hamartomas in younger children have fared poorly, including fatalities among those in the neonatal period (Hartman and Shochat 1983). Only a minority of the reported pediatric cases have been asymptomatic. Although pulmonary hamartomas typically manifest as isolated pulmonary nodules, these lesions may be quite large in young children.
Endobronchial hamartomas, which are particularly uncommon in childhood, present with respiratory symptoms or infections due to airway obstruction. A few reports of pediatric tracheal hamartomas exist, some of which may have a large extraluminal component manifesting clinically as a neck mass (Gross et al. 1996). Surgery for hamartomas is curative, although endobronchial hamartomas may require lobectomy or even pneumonectomy.
4.4 Pulmonary Chondroma
Pulmonary chondromas are benign lesions usually composed of myxoid rather than hyaline cartilage. The presence of a thin fibrous pseudocapsule, frequent bone metaplasia and calcification, and the absence of entrapped respiratory epithelium, smooth muscle, or fat allow distinction of pulmonary chondromas from hamartomas. On imaging, pulmonary chondromas appear as well-circumscribed nodules with or without calcification/ossification (Rodriguez et al. 2007). Pulmonary chondromas are the second most common component of the Carney triad, a rare nonfamilial disorder predominantly occurring in young women that also includes gastrointestinal stromal tumors (GISTs) and extraadrenal paragangliomas (Stratakis and Carney 2009) (Fig. 5). The chondromas associated with the Carney triad are usually asymptomatic and can be multiple (Rodriguez et al. 2007).
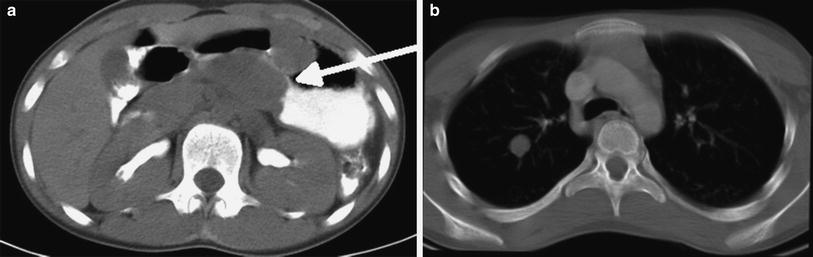
Fig. 5
Pulmonary chondroma. a Axial contrast-enhanced abdominal CT image in a 13-year-old female with incomplete Carney triad demonstrates a solid mass (arrow) along the distal wall of the stomach representing a gastrointestinal stromal tumor (GIST). b Axial chest CT image in the same patient shows a well-defined, solid right upper lobe pulmonary nodule without obvious calcification
4.5 Fetal Lung Interstitial Tumor
Fetal lung interstitial tumor (FLIT) is a recently described, very rare tumor composed of immature interstitial mesenchyme in association with irregular airspace-like structures resembling the fetal lung at 20–24 weeks of gestation. In a case series of 10 patients, all presented by 3 months of age, and two were detected by prenatal ultrasound. Other than the two cases detected antenatally, all patients presented with respiratory distress (Dishop et al. 2010). One of the patients detected prenatally presented with a large pulmonary tumor causing fetal hydrops due to inferior vena cava obstruction and had ex utero intrapartum treatment (EXIT procedure) at 37 weeks gestational age (Lazar et al. 2011).
On imaging, these tumors are well-circumscribed, unifocal, and confined to a single lobe with a predilection for the right hemithorax. A majority are solid, although some may be partially cystic. They are homogeneously low in attenuation on CT imaging and the single case evaluated by MRI demonstrated heterogeneously increased signal intensity on T2-weighted images (Fig. 6). Prognosis is favorable, with no reports of malignant transformation or recurrence, even in incompletely resected cases (Dishop et al. 2010).
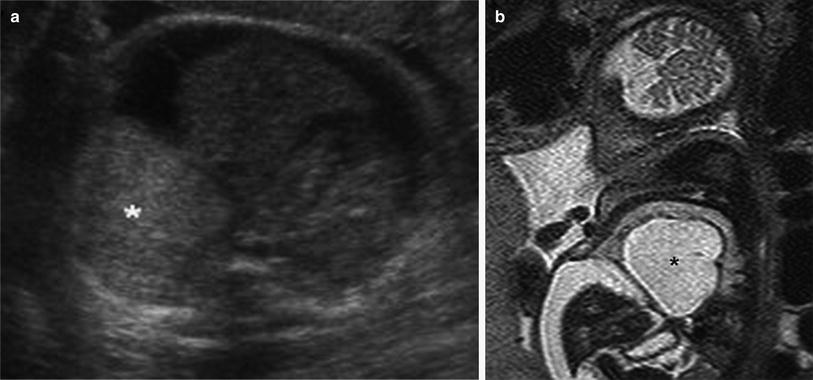
Fig. 6
Fetal lung interstitial tumor. a Transverse image from prenatal ultrasound exam performed at 36 weeks gestation reveals a large, well-circumscribed, solid hyperechoic right basilar lung mass (asterisk). b T2-weighted sagittal fetal MR image of the same patient demonstrates a large, well-circumscribed, high signal intensity mass (asterisk) compressing the adjacent lung and everting the diaphragm, with associated ascites related to inferior vena cava compression
4.6 Infantile Myofibromatosis
Infantile myofibromatosis, the most common fibrous tumorous condition of infancy, most often presents with a firm nodular mass in the subcutaneous tissues. This entity, defined by a benign proliferation of fibroblasts and myofibroblasts, is subdivided into solitary and multicentric forms that may or may not have visceral involvement (Dishop and Kuruvilla 2008). The median age of presentation is 5 months for multicentric forms and 26 months for solitary forms (Levine et al. 2012). Myofibromas usually appear as a mass with a thick-wall and a hypoechoic or anechoic center on ultrasound, and can demonstrate peripheral calcifications and adjacent bony erosion on CT. On MRI, they are usually hypointense on T1-weighted images and hyperintense with variable central hypointensity on T2-weighted images, and exhibit peripheral contrast enhancement (Koujok et al. 2005) (Fig. 7). Pulmonary masses may be a manifestation of multicentric visceral involvement and should not be misinterpreted as metastatic disease. Multicentric myofibromatosis with visceral involvement is associated with high morbidity and mortality despite chemotherapy and surgery, while the prognosis is excellent in other forms with surgery alone (Levine et al. 2012).
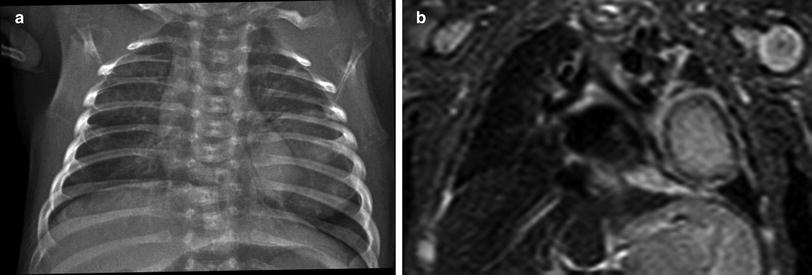
Fig. 7
Infantile myofibromatosis. a Frontal chest radiograph of a 1-month old shows a large, well-circumscribed mass of the left lower lung zone. b STIR coronal MR image of the same patient confirms the presence of a large left lung mass which exhibits central signal hyperintensity and a peripheral low signal intensity rim
4.7 Congenital Peribronchial Myofibroblastic Tumor
Congenital peribronchial myofibroblastic tumor (CPMT) is a very rare tumor that is thought to develop at approximately 12 weeks of gestational age and demonstrates variable degrees of smooth muscle and cartilaginous differentiation (Alobeid et al. 1997). The tumor is typically detected in the neonatal period, and antenatal presentation with fetal hydrops has been reported (Horikoshi et al. 2005). Although histologically benign, CPMT can exhibit certain aggressive features such as frequent mitoses, necrosis, and infiltrative growth, leading some cases to be reported as malignant entities such as bronchopulmonary fibrosarcoma (Kim et al. 2013). No syndromic, genetic, or maternal associations have been identified (Travis et al. 2004).
On imaging, CPMT typically appears as a large, single well-circumscribed pulmonary mass without lobar predilection. The tumors typically measure 5–7 cm in diameter and occupy the majority of the involved hemithorax, exerting mass affect with mediastinal shift (Travis et al. 2004). Hydrops fetalis is reported in 36 % and polyhydramnios in 27 % of cases (Horikoshi et al. 2005). CPMT has a heterogeneously hyperechoic appearance on ultrasound and a heterogeneous appearance on CT without cysts or calcifications (Travis et al. 2004). The appearance on MRI has not been described.
Patients generally survive CPMT following complete surgical resection. However, some die in the prenatal or neonatal period as a result of hydrops fetalis or respiratory failure (Huppmann et al. 2011). No local recurrence or metastasis of CPMT has been reported.
4.8 Inflammatory Myofibroblastic Tumor
Inflammatory myofibroblastic tumor (IMT) is a borderline or low-grade malignancy that can occur at a variety of sites, with the lungs being second only to the abdomen. Previously known as inflammatory pseudotumor or plasma cell granuloma, the current nomenclature emphasizes the dominant component of spindle cells, accompanied by variable numbers of inflammatory cells, particularly eosinophils, lymphocytes, and plasma cells (Yousem et al. 2004).
The important function of the myofibroblast in tissue repair is consistent with the hypothesis that an aberrant response to tissue injury underlies the pathogenesis of these lesions. A variety of unusual microorganisms have been implicated in individual case reports, including Mycobacterium avium intra–cellulare, Corynebacterium equi, Coxiella burnetii, and Bacillus sphaericus (Hedlund et al. 1999). More recently, viruses such as EBV and HHV-8 have also been implicated (Mergan et al. 2005).
IMT has been reported to account for 14 % of primary lung tumors, a similar proportion to tumors such as pleuropulmonary blastoma and bronchial carcinoid (Hancock et al. 1993). The mean age of presentation is 13 years. Although pulmonary IMT can be asymptomatic, it can also present with cough, chest pain, or a constellation of fever, weight loss, anemia, thrombocytosis, polyclonal hyperglobulinemia, and elevated inflammatory markers (Coffin et al. 2007). An association with hypertrophic pulmonary osteoarthropathy has been reported (Mas Estelles et al. 1995).
The typical imaging appearance of IMT is a well-circumscribed, solitary, upper lobe predominant pulmonary mass, with a mean size of 8.4 cm (Coffin et al. 2007; Siminovich et al. 2012). About 80–85 % is parenchymal, with the remainder being endobronchial (Hancock et al. 1993) (Fig. 8). IMTs typically are low in attenuation on noncontrast CT imaging with slight enhancement on early phase post-contrast imaging and heterogeneous enhancement on delayed phase post-contrast imaging, although single phase contrast-enhanced CT scanning should generally suffice for lesion evaluation (McHugh and Disini 2011). Calcification or cavitation is rarely seen. On MRI, IMTs typically demonstrate homogeneous slightly low signal intensity on T1-weighted images, variable hyperintensity on T2-weighted images, and heterogeneous contrast enhancement that is most pronounced in the delayed phase (Takayama et al. 2008). Hilar lymphadenopathy and pleural effusion are not commonly associated (Mas Estelles et al. 1995). Although bronchi and vessels may be trapped within the pulmonary masses and become narrowed distally or even obliterated, atelectasis is observed in only 14 % of pediatric patients (Verbeke et al. 1999).
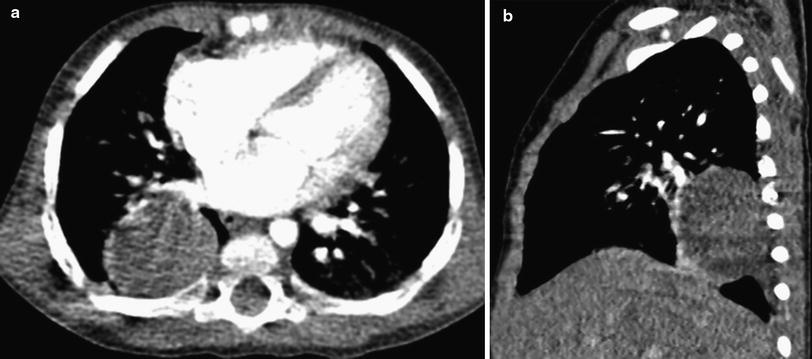
Fig. 8
Inflammatory myofibroblastic tumor (IMT). a Axial contrast-enhanced chest CT image in a 13-month-old girl who had intermittent unexplained fever. A chest radiograph had shown a rounded right lower lobe mass, confirmed to be solid on CT. b Sagittal contrast-enhanced CT image shows pleural extension of the mass posteroinferiorly, but no associated pleural effusion or chest wall invasion
Locally aggressive IMTs are occasionally seen and may invade the mediastinum, chest wall, or diaphragm (Hedlund et al. 1999; Verbeke et al. 1999; Yousem et al. 2004). CT and MRI are both useful in characterizing the extent of these aggressive lesions in relation to the airways, esophagus, cardiovascular structures, and chest wall (Hedlund et al. 1999). Lesions arising solely in the esophagus have been described, and barium esophagography can be useful for characterizing esophageal involvement when suspected.
Over-expression of the ALK1 (anaplastic lymphoma kinase) gene is associated with an increased rate of local recurrence but not distant metastases, and an overall improved prognosis. Recurrent and metastatic IMTs tend to be larger (Coffin et al. 2007). Many IMTs act in a relatively indolent fashion, although about one-quarter recur. Pulmonary resection with removal of all gross evidence of disease is the mainstay of treatment and is usually curative for tumors confined to the lung. Locally aggressive lesions may require more radical surgery including pneumonectomy rather than the more usual segmental or lobar resection. When these tumors extend beyond the organ of origin at diagnosis, up to 46 % relapse locally (Janik et al. 2003). Relapse or invasion of the mediastinum has occasionally been treated with immunosuppressive therapy or even multiagent chemotherapy with good results (Verbeke et al. 1999; Janik et al. 2003). Metastases have been documented to the lung, liver, bone, and brain (Coffin et al. 2007; Siminovich et al. 2012).
4.9 Pleuropulmonary Blastoma
Although previous reports report a similar incidence to IMTs and bronchial carcinoids (Hancock et al. 1993), pleuropulmonary blastoma (PPB) is likely the most common primary lung malignancy of childhood (Priest et al. 1997). The mean age at presentation is 38 months, and it is rarely seen in children over 6 years of age. Prenatal presentation has also been reported (Miniati et al. 2006). Approximately 30–40 % of PPB patients or their relatives have manifestations of the PPB Family Tumor Dysplasia Syndrome (PPB-FTDS), an autosomal dominant genetic disorder with a distinctive predisposition to certain tumors and dysplasias, including PPB, cystic nephroma, multinodular goiter, Sertoli-Leydig cell tumor, cervical embryonal rhabdomyosarcoma, pituitary blastoma, pineoblastoma, nasal chondromesenchymal hamartoma, and ciliary body medulloepithelioma (Slade et al. 2010; Foulkes et al. 2011). Approximately 70–75 % of patients with PPB have germline mutations in the DICER1 gene that encodes a protein involved in microRNA processing (Priest 2012).
PPB is composed of a malignant mesenchymal component resembling fetal lung in early gestation with a variably sarcomatous or blastematous appearance, but no malignant epithelial component. Rhabdomyosarcomatous areas in PPB are identical to embryonal rhabdomyosarcoma, and cases of PPB have been misdiagnosed as rhabdomyosarcomas arising in a congenital cystic adenomatoid malformation (CCAM) (Pai et al. 2007) or in normal lung (Schiavetti et al. 2009). Recognition of high grade blastemal elements, cartilage, spindle cell sarcoma, or diffuse-marked anaplasia facilitates the correct diagnosis of PPB in a pediatric lung mass. PPB is distinct from pulmonary blastoma, a biphasic sarcomatoid carcinoma predominantly occurring in adults (Dehner 1994).
Three subtypes of PPB are described based on gross morphology. Type I is a unilocular or multilocular cyst with delicate fibrous septa, Type II PPB is mixed cystic and solid and Type III PPB is entirely solid (Dehner et al. 1995; Priest et al. 2006). There is a significant difference in the age at presentation. The median age at diagnosis is 10 months for type I, 34 months for type II, and 44 months for type III (Priest et al. 2006). Progression of unresected Type I PPB to Type II and III PPB can occur. Type I PPB may also spontaneously regress, whereby it is classified as Type I-regressed PPB (Type Ir PPB). Types I and Ir PPB are indistinguishable on gross inspection from the large-cyst form of CCAM; however, CCAMs do not transform into PPBs (Priest et al. 2009).
The clinical presentation of PPB is varied and nonspecific, ranging from incidental detection to cough, chest pain, and respiratory distress. Type I and Ir PPBs appear as well-defined, air-filled cysts confined to the lung parenchyma or visceral pleura, and the presence of multifocal cysts or spontaneous pneumothorax favors type I PPB over large-cyst CCAM (Priest et al. 2009). Type II and III PPBs typically manifest as a large mass in the hemithorax, often in the lung periphery adjacent to the pleura, with associated mediastinal mass effect and pleural effusion (Priest et al. 2006) (Fig. 9). Invasion of the chest wall or diaphragm is occasionally observed. Local invasion of the bronchi, great vessels, and heart is rare (Goel et al. 2010; Priest et al. 2011). The solid components of the mass demonstrate contrast enhancement on CT and MRI. Some type II and III PPBs are sharply demarcated from adjacent lung parenchyma while others may be more infiltrative. Confident assignment of the site of origin is often difficult to determine with large lesions.
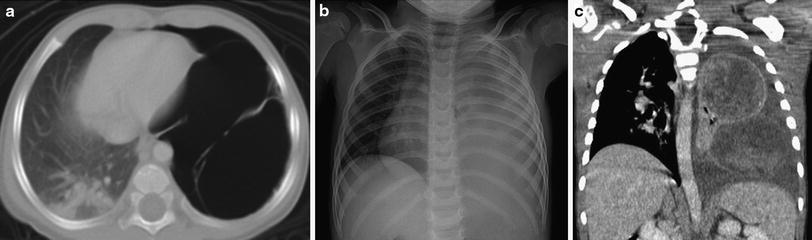
Fig. 9
Pleuropulmonary blastoma (PPB). a Axial chest CT image from a 1-year old with a history of spontaneous pneumothorax demonstrates a large, thin-walled, septated, air-filled, cystic lesion of the left lung representing a Type I (cystic) PPB. b Frontal chest radiograph of a different patient at 3 years of age shows near complete opacification of the left hemithorax with rightward mediastinal shift, findings evocative of complicated pneumonia with empyema. c Coronal contrast-enhanced chest CT image reveals a bilobed heterogeneously enhancing mass arising from the left lung representing a Type III (solid PPB), with an associated left pleural effusion
Initial misdiagnosis is common, with Type I PPBs mimicking large-cyst CCAMs, and type II and III PPBs mimicking other tumors or complicated pneumonia with empyema. These tumors are so friable intraoperatively that empyema may still be suspected during surgery (Buyukavci et al. 2006). The diagnosis depends on histological evaluation that in most cases takes place after attempted or successful surgical resection. The malignant elements in Type I PPBs can be very subtle, requiring meticulous inspection of the pathology specimen to make the correct diagnosis.
Type I PPBs have a better prognosis than the other subtypes. Local recurrence is unusual in Type I PPB, occurring in fewer than 15 %, but develops in more than 45 % of Types II and III. Metastases occur in 11 % of Type II and 55 % of Type III cases (Priest 2012). Metastases have a particular tropism for the central nervous system including the spinal cord (Priest et al. 1997), so that craniospinal MRI is merited at staging and follow-up. The second most common site for metastatic spread is the skeletal system. Metastases to the liver, adrenals, and ovary are also reported (Priest 2012). The use of adjuvant chemotherapy in addition to surgery confers a survival advantage for all three subtypes (Priest et al. 2006). Overall survival is 90 % for Type I and 40–60 % for Types II and III (Priest 2012). Extrapulmonary involvement and incomplete resection confer a worse prognosis (Indolfi et al. 2007).
4.10 Bronchial Carcinoid Tumor
Bronchial carcinoid tumors arise from neuroendocrine Kulchitsky cells in the airways. Although bronchial carcinoid is the most common malignant endobronchial tumor and the second most common primary pediatric malignancy of the lower respiratory tract (Dishop and Kuruvilla 2008), it is still uncommon in children relative to adults. When it occurs in children, it tends to present in adolescents (Al-Qahtani et al. 2003). The actual carcinoid syndrome is very rare in childhood outside the setting of metastatic disease (Hancock et al. 1993). Cushing syndrome from ectopic adrenocorticotropic hormone secretion has also been described but is similarly uncommon (Wang et al. 1993). Children with bronchial carcinoids are much more likely than their adult counterparts to present with post-obstructive wheezing and atelectasis, in addition to hemoptysis and pneumonitis (Wang et al. 1993). A carcinoid in an asymptomatic patient is more likely to involve a peripheral airway than a central airway (Curtis et al. 1998). Bronchial carcinoids usually have intraluminal, mural, and extrabronchial components. Up to 4 % of carcinoid tumors are associated with other endocrine neoplasias, the most common association being with pituitary tumors (Al-Qahtani et al. 2003).
The majority of carcinoid tumors are not visible on CXR and their true extent may be difficult to determine even on high-resolution CT. The typical CT appearance is of a single round or lobulated mass that is at least partially endobronchial (Jeung et al. 2002). They are hypervascular polypoid tumors and characteristically show prominent contrast enhancement. Occasionally, the mass external to the bronchus is larger than that in the lumen, and the extrabronchial component may consequently be visible as a hilar mass. The mass resides along the main, lobar, or segmental bronchi in 80 % of cases, with a predilection for branching sites. The mass involves the more peripheral airways in the other 20 % of cases (Chong et al. 2006). Approximately one quarter of carcinoid tumors in adult patients are calcified, but the frequency of calcification is much less in childhood. Bronchial carcinoids typically demonstrate hyperintensity on T2-weighted images and avid homogeneous contrast enhancement on MRI. Bronchial carcinoids show increased octreotide uptake on somatostatin receptor scintigraphy in greater than 85 % of cases (Jeung et al. 2002) (Fig. 10). Although not yet widely available, the novel PET tracers 68-Ga-DOTATOC and 18F-DOPA in combination with integrated CT offer higher spatial resolution than conventional somatostatin scintigraphy and improved sensitivity for the detection of carcinoid tumor (Koopmans et al. 2008; Buchmann et al. 2007). The incidence of metastases with bronchial carcinoids in children is 10–15 % (Wang et al. 1993). Metastases are most commonly seen in the liver, adrenals, brain, and bone. Pediatric bronchial carcinoids generally have an excellent prognosis with complete resection and lymph node excision. Recurrence has been reported in as many as 20 % of cases but usually can be successfully treated surgically (Rizzardi et al. 2009).