Imaging the Pediatric Brain
The central nervous system (CNS) consists of the skull, brain, spine, and spinal cord. Imaging the brain in children differs from imaging adult patients in many respects. Ultrasound, a modality rarely used in adult neuroradiology, plays a central role in imaging the brain in fetal life and infancy. Similar to adult neuroradiology, magnetic resonance imaging (MRI) is now the most widely used modality to image the brain in older children. In addition, MRI is of particular benefit in pediatric patients given a lack of ionizing radiation. Also, many advanced MRI techniques have important and unique applications in pediatric practice. Computed tomography (CT) continues to play an important role in the rapid evaluation of acute trauma and evaluation of the bony calvarium, but its use for other indications in pediatric patients has diminished as pediatric imagers, clinicians, and families strive to reduce exposure to ionizing radiation in children. Positron emission tomography (PET) allows functional imaging based on glucose metabolism, and the growing use of PET-CT and PET-MRI allows the coregistration of metabolic and structural information, which is especially important in epilepsy imaging. The use of catheter angiography for diagnostic purposes has certainly diminished with advances in noninvasive CT and magnetic resonance angiography (MRA) techniques, but catheter angiography maintains a unique role in diagnostic imaging, and the number of types of catheter-based interventions in pediatric neuroradiology continues to increase.
Radiography
Skull radiographs lack both sensitivity and specificity for most clinical indications and carry a not insignificant radiation dose. Their use has fallen with the increased utilization of cross-sectional imaging. Skull radiographs have a limited role in the evaluation of trauma or suspected skull fracture where, in general, if imaging is deemed clinically appropriate, CT is preferred. However, skull radiographs continue to be included in skeletal surveys performed for suspected child abuse. The use of radiographs in the evaluation of craniosynostosis has largely been superseded by CT. The use of skull radiographs is no longer considered routine in the evaluation of a palpable head lump, with most practitioners preferring the use of ultrasound and/or CT-MRI.
Ultrasound
Ultrasound is the primary modality used for imaging the fetal and neonatal brain. Screening fetal ultrasound can detect many structural brain anomalies in utero, which can be further assessed with more detailed ultrasound scanning and/or fetal MRI.
After birth, the open anterior and posterior fontanels provide acoustic windows that allow exquisite depiction of the infant brain by ultrasound. Neonatal head ultrasound was first performed in the 1970s, but the image quality has improved immensely in recent years with the development of better high-frequency transducers with smaller footprints. Head ultrasound is now the most commonly used neuroimaging modality in neonates.
Advantages of ultrasound include accessibility, portability, rapid image acquisition (obviating the need for sedation), and low cost. The use of head ultrasound is particularly important when imaging critically unwell newborns. Ultrasound can be performed portably in the neonatal intensive care unit providing important real-time diagnostic information, which is especially important with neonates who are too unstable to be transferred or for neonates receiving extracorporeal membrane oxygenation. Ultrasound can be used to screen for congenital anomalies, hydrocephalus, intracranial hemorrhage, periventricular leukomalacia, and other abnormalities. Disadvantages of ultrasound are its limited sensitivity for ischemia (especially in the early stages of ischemic injury) and sometimes limited views of the peripheral structures and posterior fossa.
American College of Radiology guidelines for a standard head ultrasound consist of a series of sagittal, parasagittal, and coronal grayscale images acquired through the anterior fontanel. Ultrasound is operator dependent, and it is important to adjust time gain compensation (TGC) curves and to use multiple focal zones to optimize the imaging during acquisition. A combination of vector, curved, and linear array transducers can be used to produce diagnostic images of both superficial and deeper structures. Color and spectral Doppler can be performed to assess the circle of Willis, the cortical veins and venous sinuses, and suspected vascular anomalies such as vein of Galen malformations. Supplemental images of the posterior fossa can be acquired via the mastoid fontanel, and imaging of the craniocervical junction can be achieved by using the “foramen magnum view,” which involves scanning below the occipital protuberance. In addition to static images, real-time cine sweeps can be useful. Head ultrasound becomes progressively more technically challenging as the anterior fontanel closes, and it is rarely of diagnostic quality in children older than 9 months.
Magnetic Resonance Imaging
For the majority of clinical indications in most children, MRI is the modality of choice when imaging of the brain is required. MRI has many advantages over other modalities including excellent soft tissue resolution, excellent spatial resolution, multiplanar imaging acquisition, and very importantly in pediatric patients it does not involve the use of ionizing radiation. One of the main disadvantages of MRI is the longer time required to obtain a diagnostic study compared with other modalities. MRI is also very susceptible to imaging artefact related to patient motion. For these reasons, pediatric MRI studies are often performed using sedation or general anesthesia. In general, neonates and infants younger than 6 months do not require sedation. An infant who is recently fed, swaddled, warmed, and has hearing protection in place will often tolerate a lengthy MRI examination without sedation. Between the ages of 6 months and 4 years, it is usually necessary for children to be sedated for MRI. Between the ages of 4 and 6 years, many, but not all, children can cooperate with an MRI examination. Most children older than 6 years will cooperate with MRI without sedation. Child life specialists and distraction techniques such as the use of video goggles and an entertaining movie can be invaluable in achieving a diagnostic study without sedation.
The individual sequences chosen for an MRI protocol vary depending on the clinical indication, the age of the patient, and the individual scanner. In general, an MRI of brain includes sagittal T1-weighted imaging, axial and coronal T2-weighted imaging, axial T2 fluid attenuation inversion recovery (FLAIR), and axial diffusion-weighted imaging (DWI). A brain MRI does not always require the administration of a paramagnetic contrast agent such as gadolinium; however, postcontrast imaging can be used, in particular for the identification and characterization of primary and metastatic brain tumors and leptomeningeal disease, intracranial infections, demyelination, and neurocutaneous disorders. Administration of gadolinium can also allow the assessment of cerebral perfusion using dynamic susceptibility contrast MRI techniques.
DWI uses gradient sequences to generate images based on differences in the rate of diffusion of water molecules. The rate of diffusion, or apparent diffusion coefficient (ADC), is higher for free water (eg, cerebrospinal fluid [CSF]) than for macromolecular-bound water (eg, gray matter and white matter). DWI is very sensitive to primary or secondary derangements of cellular energy metabolism such as hypoxia and ischemia, hypoglycemia, inborn errors of metabolism disorders, viral encephalitis, and status epilepticus. These changes are manifest as reduced ADC (ie, high intensity on DWI and low intensity on ADC maps). Acute diffusion restriction can be seen within minutes of injury and much earlier than on conventional imaging sequences. Current clinical applications of DWI include the assessment of brain maturation, the evaluation of ischemia, and the characterization of tumors. Diffusion tensor imaging (DTI) is a technique whereby characterization of the magnitude, anisotropy, and orientation of the diffusion tensor allows the creation of three-dimensional (3D) tractographic maps of the white matter; these can be invaluable when planning surgery for pediatric patients.
Gradient recalled echo (GRE) sequences can be used to generate susceptibility-weighted images (SWI); the signal void caused by magnetic susceptibility artefact may be the only evidence of old hemorrhage and can be especially useful in the investigation of traumatic brain injury (TBI) and concussion.
Two-dimensional and 3D time-of-flight (TOF) MRA techniques use flow compensation gradients and permit the creation of superb maximal intensity projection angiograms and venograms based on flow relating signal without the need for administration of intravenous contrast.
CSF flow dynamics are often of particular interest in pediatric patients, for example, in the setting of hydrocephalus, Chiari malformation, or evaluating response to treatments such as endoscopic third ventriculostomy (ETV). Phase-contrast techniques can provide both quantitative and qualitative information regarding CSF flow. Qualitative information can be obtained from the more widely used steady-state free procession techniques (eg, constructive interference in steady state (CISS), fast imaging with steady-state precession (FISP), steady state free precession (SSFP)).
Arterial spin labeling is an MRI technique that uses a radiofrequency pulse to magnetically “tag” arterial blood water before it enters the tissue of interest. Using a delay between tagging and image acquisition allows quantitative and qualitative measurement of tissue perfusion. This technique can be used in ischemic injury, epilepsy, moyamoya, and tumor evaluation. It is especially important in pediatric patients because the CT perfusion techniques widely used in adult practice are associated with a high radiation dose.
Proton (hydrogen) magnetic resonance spectroscopy (MRS) allows noninvasive assessment of cellular metabolism, and this is especially important when evaluating newborns with hypoxic ischemic encephalopathy, children with suspected neurometabolic disorders, and children with brain tumors. MRS can provide quantitative information regarding cellular metabolites before the detection of morphological changes by MRI or other imaging modalities. The proton MR spectrum in normal tissue is characterized by at least three peaks. The dominant peak corresponds to N -acetyl aspartate (NAA), which is a marker of neuronal integrity/density. The peak associated with creatine and phosphocreatine represents cellular energy metabolism, and the peak associated with choline represents cell membrane synthesis. A lactate peak is not seen in the normal spectrum, but a characteristic lactate doublet can be demonstrated with inflammation, infarction, and some tumors.
Blood oxygen level–dependent contrast imaging uses the T2* effect of deoxyhemoglobin to identify focal areas of increased perfusion in the brain during the performance of specific tasks. This functional MRI (fMRI) technique has been shown to localize the motor strip, speech center, and memory centers. fMRI is an area of intense ongoing research, but it has yet to translate into widespread clinical practice.
Computed Tomography
Modern, multidetector CT scanners permit rapid, high-resolution imaging acquisition in the axial plane and creation of isotropic, multiplanar, reformatted images, or 3D reconstructions using a variety of soft tissue, bone, vascular, and other algorithms. The speed of acquisition is one of the main advantages of CT, particularly in the setting of trauma where prompt diagnosis is critically important. CT is sensitive at depicting skull fractures, pneumocephalus, acute intraaxial or extraaxial hemorrhage, and herniation. Another indication where CT is preferred to other modalities is the evaluation of children with craniofacial anomalies or craniosynostosis.
The benefits of CT need to be weighed against the risks related to the use of ionizing radiation. When performing CT in pediatric patients, it is important to collimate appropriately to the area of interest and use child- and indication-appropriate parameters with reduced kilovoltage (kV) and milliampere-second (mAs). The increased use of more rapid iterative reconstruction techniques on modern clinical scanners has allowed further reductions in radiation dose.
It is especially important to reduce lifetime radiation exposure in children with chronic health conditions who may undergo many diagnostic imaging investigations in their lifetime. In the past, many children with ventriculoperitoneal shunts received a large number of CT examinations for the evaluation of suspected shunt malfunction. Many centers now use a truncated MR protocol consisting of a rapidly acquired axial steady-state free procession sequence to screen for signs of shunt malfunction in preference to performing a head CT. If a rapid MRI is not available in this setting, a lower-dose CT may suffice to evaluate the size of the ventricles.
Contrast-enhanced CTA can provide exquisite vascular imaging. A tight bolus of contrast (3 mL/kg body weight up to a total dose of 120 mL) should be administered through a large-bore intravenous catheter to achieve a good-quality CTA. Adverse reactions to contrast are rare in pediatric patients; asthma and previous reactions to contrast medium are risk factors for acute reaction. Advantages of CTA over MRA include more rapid acquisition and better spatial resolution, but given the high radiation dose involved, the technique is less used in pediatric patients than in adults.
Positron Emission Tomography
PET imaging is based on the detection of photons related to the decay of injected radiotracers. 18-Fluorodeoyglucose (FDG), which is a marker for glucose metabolism, is the most widely used tracer in current clinical practice. Interictal FDG-PET studies may show hypometabolism corresponding with an epileptogenic focus which may not be apparent on conventional imaging. Coregistration with CT or MRI can improve the presurgical workup in patients with intractable seizures. Currently, FDG-PET has a limited part in the evaluation of pediatric brain tumors, but there is ongoing research with newer tracers such as labeled amino acids, which may be able to provide information regarding presence or absence of tumor, grade of tumor, and treatment response and distinguish between tumor recurrence and radiation necrosis in the future.
Congenital Malformations of Brain Development
Malformations of brain development may be due to a variety of causes, including genetic abnormalities, infections, toxins, and ischemia. Recent advances in genetics, molecular biology, and imaging have resulted in an improved understanding of the pathogenesis and manifestations of many malformations of the CNS. As our knowledge of malformations has improved, the classification of the various malformations has evolved. In the paragraphs that follow, we will be referencing the classification scheme updated by Barkovich and colleagues in 2012 (see Suggested Readings for more details).
Malformations of Cortical Development
The cerebral cortex is the outermost layer of gray matter that covers the cerebrum. Histologically it is divided into six layers, each with a characteristic distribution of neuronal cell types and connections with other cortical and subcortical regions. Embryologically, the cortical neuronal cells originate as progenitor cells in the periventricular germinal matrix. These progenitor cells differentiate into neurons and glial cells. The neurons start to proliferate around the seventh week of gestation and begin their migration from the periventricular zone to the surface of the brain along radially oriented glial fibers around the eighth week. After migration, there is progressive organization of the cortex with refinement of connectivity between neurons as development proceeds. Disruption of any of the normal developmental processes can result in a malformation of the cortex. Although the phenotype of many malformations is determined by multiple errors along the pathway of normal development, the malformations are currently classified based on the earliest process that is interrupted: proliferation, migration, or organization [ Table 8.1 ]. Note that in this classification system, the term cortical dysplasia is a specific entity and the term should not be used in a general fashion to describe other malformations of cortical development.
| Disorders of Proliferation | Disorders of Migration | Disorders of Organization |
|---|---|---|
|
|
|
Disorders of Neuronal Proliferation
Microlissencephaly.
Microlissencephaly is used to describe children with severe microcephaly, a simplified sulcal pattern, and a thickened cortex. Different causative factors have been implicated including cytomegalovirus (CMV) infection and mutations of the RELN gene. Children with microlissencephaly tend to present with seizures and profound developmental delay.
Hemimegalencephaly.
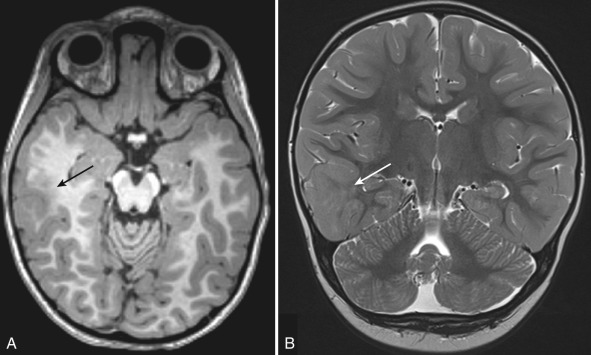
Hemimegalencephaly refers to hamartomatous overgrowth of all or part of a cerebral hemisphere. Hemimegalencephaly refers to a heterogeneous group of conditions and may occur in isolation or in association with hemihypertrophy and neurocutaneous disorders. In some cases, there is a familial predisposition.
Children with hemimegalencephaly may present with macrocephaly, hemiplegia, seizure disorder, and developmental delay. MRI reveals diffuse enlargement of an entire cerebral hemisphere or a specific lobe of the brain [ Fig. 8.1 ]. Other anomalies such as polymicrogyria (PMG), lissencephaly, or gray matter heterotopia may be present. The ipsilateral lateral ventricle tends to be enlarged. In patients with intractable seizures, anatomic or functional hemispherectomy may be performed. Surgery may be contraindicated if there are contralateral malformations. Therefore a careful assessment of the contralateral “normal” hemisphere is especially important when reviewing MRI studies in these patients.

Focal Cortical Dysplasias.
Focal cortical dysplasias (FCD) are not associated with a single, known genetic abnormality and most likely arise because of mutations in a number of different genes. Histologically, FCD is divided into three main categories based on cortical delamination, disruption of cell architecture, cell composition, and any associated destructive brain lesions. The most common clinical presentation of children with FCD is medication-refractory, focal epilepsy.
Identification of FCD on MRI can be challenging. High-resolution T1, T2, and FLAIR images need to be scrutinized carefully for subtle cortical thickening with or without T1 shortening, abnormal gyration with an indistinct interface between the cortex and subcortical white matter, and the presence of adjacent white matter signal abnormality [ Fig. 8.2 ]. The transmantle sign has been described in type II FCD and refers to T2 prolongation (hyperintensity) within the subcortical white matter underlying the cortical abnormality extending to the ventricle. The signal abnormality is due to the presence of balloon cells and ectopic neurons along the radial glial pathway. Because MR delineation of a lesion for resection strongly correlates with surgical outcome, identification of FCD is critically important in patients, with intractable epilepsy being considered for operative intervention. FCD can be clinically occult and is occasionally seen on MRIs performed for an indication other than seizure. The differential diagnosis for an FCD on MRI often includes low-grade neoplasm. Follow-up imaging to assess stability of the lesion may help differentiate FCD, which should not change over time, from a slowly growing neoplasm.
Disorders of Neuronal Migration
Lissencephaly.
Lissencephaly means “smooth brain” and is characterized by reduced gyral and sulcal development. It is also referred to as the agyria-pachygyria complex. Agyria is defined as absence of the gyri in association with a thickened cortex; pachygyria is defined as the presence of a few broad, flat gyri. Lissencephaly encompasses a number of heterogeneous disorders that can be grouped into two major types: type 1 (classic) lissencephaly and type 2 (cobblestone lissencephaly).
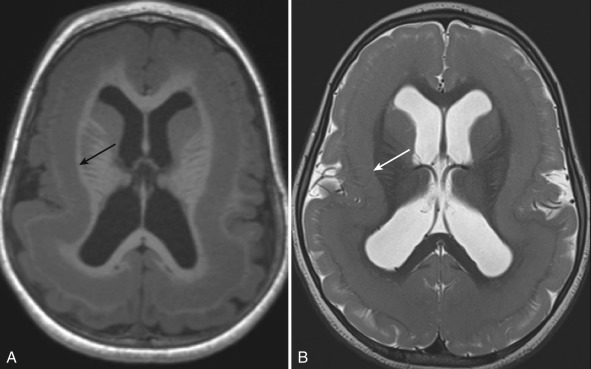
Type 1 (classic) lissencephaly occurs in 1 in 500,000 live births and is caused by disruption of neuronal migration between the 10th and 14th weeks of gestation. Up to 70% of patients with type 1 lissencephaly have mutations of either LIS1 or doublecortin ( DCX ) gene. LIS1 type 1 lissencephaly includes the Miller-Dieker syndrome and results from a de novo mutation in 80% of affected children. DCX has an X-linked pattern of inheritance. Males with the DCX mutation have lissencephaly, whereas heterozygous females have subcortical laminar heterotopia referred to as “band heterotopia.”
Children with type 1 lissencephaly are typically hypotonic at birth with a normal or small head circumference. They subsequently experience development of seizures, hypertonia, and hyperreflexia. On imaging, type 1 lissencephaly consists of a diffusely thickened and abnormally smooth cerebral cortex. Band heterotopia or “double cortex” is characterized by a layer of incompletely migrated neurons and more normally gyrated appearance of the outer cortex [ Fig. 8.3 ]. The clinical phenotype tends to be much less severe in band heterotopia.
Type 2 (cobblestone) lissencephaly occurs because of excessive neuronal migration. It represents a heterogeneous group of disorders characterized by morphological abnormalities in the brain and merosin-negative or merosin-deficient congenital muscular dystrophy including Fukuyama disease, muscle-eye-brain disease, and Walker-Warburg syndrome. A combination of clinical findings, elevated serum creatine kinase levels, brain imaging, muscle and skin biopsy, and molecular genetic testing helps make the specific diagnosis. The imaging appearance on MRI can vary from mild pachygyria and a lack of normal sulcation to a highly abnormal, “cobblestone” appearance to the cortex, particularly anteriorly [ Fig. 8.4 ]. Because of overmigration of neurons, the CSF spaces surrounding the brain are typically reduced. The reduction in CSF surrounding the brain can be one of the earliest signs of cobblestone lissencephaly on fetal MR. Abnormalities can also be seen in the brainstem and in the cerebellum, and occipital cephalocele may be present in Walker-Warburg syndrome.

Gray Matter Heterotopia.
Gray matter heterotopia are focal collections of gray matter that arise because of interruption of normal neuronal migration. Heterotopia can be seen in isolation or in association with other anomalies. They can be subdivided into three groups: periventricular/subependymal, focal subcortical, and leptomeningeal; periventricular heterotopia are the most common. The majority of cases are sporadic. An X-linked dominant filamin-1 gene mutation is associated with extensive periventricular heterotopia and is seen mostly in girls. Heterotopia may cause seizures; with seizures usually more severe in children with multiple heterotopia. On all imaging modalities, heterotopia appear as nodules with the same imaging features as gray matter [ Fig. 8.5 ]. fMRI and blood oxygenation level–dependent imaging can demonstrate activation in epileptogenic heterotopia. Contrast enhancement should not be present.

Disorders of Neuronal Organization
Polymicrogyria.
PMG occurs because of disruption of late neuronal migration and cortical organization. It results in abnormalities in the deeper cortical cell layers and the formation of small and overly convoluted gyri. PMG can be unilateral or bilateral and focal or diffuse. The peri-Sylvian region is the most commonly involved [ Fig. 8.6 ]. Patients can present with developmental delay, focal neurological signs/symptoms, or seizures depending on the cortical area involved. PMG can be sporadic and isolated, associated with other genetic syndromes or related to CMV infection or cerebral ischemia.

The most constant feature of PMG is cortical thickening. High-resolution imaging better depicts the small abnormal gyri and bumpy contour. A volumetric acquisition with multiplanar evaluation is often helpful to diagnose subtle irregularities of the gray-white matter junction, which may be the only evidence of PMG. In unmyelinated areas, the abnormal cortex may appear thin and bumpy, as opposed to myelinated areas where it can appear thicker and relatively smooth. Perisylvian PMG is often accompanied by an abnormally vertical orientation of the Sylvian fissure [see Fig. 8.8 ]. The abnormality of the Sylvian fissure may be one of the first clues to the presence of PMG on fetal MR. Subtle cases of PMG may be difficult to diagnose in the neonate because of the high water content in the inner cell layers of the cortex, and follow-up imaging later in infancy may be required to detect or confirm PMG.
Schizencephaly.
Schizencephaly refers to congenital clefts in the cerebral hemisphere extending from the ventricular ependyma to the pia mater overlying the cerebral cortex. Schizencephaly is a disorder of neuronal organization and the clefts are lined with heterotopic gray matter, usually PMG. This is in contrast with porencephaly, which is a cystic lesion lined by white matter that occurs secondary to an encephaloclastic insult such as trauma or infarction.
The incidence of schizencephaly is estimated at 1.5 in 100,000 live births. Patients may be asymptomatic but usually present with seizures or developmental delay.
MRI is the imaging modality of choice. The lips (margins) of the cleft may be in apposition (closed-lip) or separated (open-lip). Closed-lip schizencephaly produces a nipple-like outpouching at the ependymal surface [ Fig. 8.7 ]. In open-lip schizencephaly, the CSF cleft can be seen extending from ventricular ependyma to the cortical surface. Schizencephaly may be associated with absence of the septum pellucidum and small optic nerves in patients with septooptic dysplasia (SOD).

Abnormalities of Ventral Induction
Holoprosencephaly
Holoprosencephaly (HPE) is the most common congenital anomaly affecting the ventral forebrain. Incomplete cleavage of the forebrain, absence of the interhemispheric falx, and fusion of the central gray nuclei characterize HPE. Abnormalities of the corpus callosum are usually present in all but the most subtle forms. Because the corpus callosum normally forms in an anterior-to-posterior direction, most callosal dysgenesis is accompanied by deficiencies in the posterior portions of the corpus callosum, but in HPE, the callosal anomalies are more often seen in the anterior portion of the commissure. In HPE, a failure of hemispheric cleavage occurs during the fifth and sixth weeks of gestation. HPE occurs in 1 in 250 embryos and in 1 in 8300 to 16,000 live births. It is associated with facial anomalies including cyclopia, ethmocephaly, cebocephaly, and hypotelorism. A central “megaincisor” may be present. The more severe the facial anomalies, the more severe the brain anomalies are likely to be, leading to the statement that, in HPE, the “face predicts the brain.”
In the most severe form, known as alobar HPE, there is complete fusion of the cerebral hemispheres, with absence of the falx, corpus callosum, and septum pellucidum. A rudimentary monoventricle is present, which is often in communication with a dorsal cyst [ Fig. 8.8 ].

In semilobar HPE, the posterior parts of the interhemispheric fissure and falx are present with fusion of hypoplastic frontal lobes. The globus pallidi may be hypoplastic or absent, and the caudate nuclei and thalami are fused. The hippocampus is usually present but often dysplastic. A dorsal cyst may be present. Facial anomalies are less severe than in the alobar form [ Fig. 8.9 ].

In an unusual middle hemispheric variant of HPE, also known as syntelencephaly, the hemispheres are cleaved anteriorly and posteriorly but fused in the posterior frontal region. Syntelencephaly is not associated with craniofacial anomalies. Unlike other forms of HPE which have been linked to abnormalities in the sonic hedgehog gene, the interhemispheric variant has been linked to abnormalities in the ZIC2 gene.
A milder form, lobar HPE, is characterized by hypoplasia of the frontal poles with agenesis of the septum pellucidum. The posterior frontal, parietal, and occipital lobes are normally formed. The splenium and body of the corpus callosum are present. Usually very mild or no facial anomalies are associated with lobar HPE [ Fig. 8.10 ].

Septooptic Dysplasia
SOD was originally described by de Morsier in 1956 as hypoplasia of the optic nerves and absence of the septum pellucidum. It is now used to describe any combination of optic nerve hypoplasia, pituitary hypoplasia, and midline ventral cerebral abnormalities and is often considered a mild form of HPE. Two-thirds of patients have hypothalamic-pituitary dysfunction. The incidence is estimated at 1 in 50,000 live births. A number of different genetic mechanisms have been implicated.
Absence of the septal leaflets results in downward displacement of the fornices into the third ventricle. The posterior pituitary gland may be ectopic or absent, and the infundibulum may be interrupted. The optic nerves and chiasm show varying degrees of hypoplasia [ Fig. 8.11 ]. SOD is often associated with schizencephaly.

Anomalies of Corpus Callosum
The corpus callosum is the largest of the telencephalic commissures. Agenesis or partial absence of the corpus callosum is one of the most common congenital CNS anomalies with an incidence of 1 in 4000.
The development of the corpus callosum occurs between the 12th and 20th weeks of gestation. The genu is the first portion to form with development continuing posteriorly along the body to the splenium. The rostrum is the last portion to be formed. Agenesis is used to describe a primary failure to form the corpus callosum [ Fig. 8.12 ]. In agenesis, other interhemispheric commissures, such as the anterior commissure, are usually enlarged. Complete or partial agenesis should be distinguished from secondary callosal destruction that may be caused by toxic, ischemic, or traumatic insults.

In complete callosal agenesis, the lateral ventricles have a characteristic parallel orientation with dilatation of the trigones and occipital horns ( colpocephaly ), which has also been described as the “race-car sign” on axial imaging with the dilated occipital horns resembling the rear tires of an open-wheel car [ Fig. 8.13 ]. Axons that fail to cross the midline course along the medial aspect of the lateral ventricle forming the longitudinally oriented Probst bundles, which can be elegantly illustrated as anteroposterior tracts on DTI and as dark, densely myelinated structures along the medial wall of the lateral ventricles on T2-weighted imaging [ Fig. 8.14 ]. Callosal agenesis is often associated with an interhemispheric cyst and a high riding third ventricle. On sagittal imaging, there is absence of the cingulate gyrus with a radial orientation of the sulci over the mesial surface of the frontal lobes.


Callosal agenesis may be isolated or associated with a multitude of other malformations including Chiari II malformation, aqueductal stenosis, and anomalies of cortical, cerebellar, and brainstem development. Aicardi syndrome is a rare X-linked dominant condition that occurs in females who present with seizures and developmental delay. Imaging findings in Aicardi syndrome include callosal agenesis, interhemispheric neuroepithelial cysts, gray matter heterotopia, PMG, cerebellar malformation, spinal anomalies, chorioretinal lacunae, and coloboma [ Fig. 8.15 ].

Partial callosal agenesis is usually characterized by absence of the splenium and dorsal body with preservation of the genu and rostral body. Diffuse callosal hypoplasia and segmental callosal hypoplasia refer to global and focal thinning of the callosum, respectively.
Intracranial lipomas indicate abnormal brain development and are usually associated with hypoplasia of the adjacent brain. Pericallosal lipomas are usually associated with partial absence or formational anomalies of the corpus callosum. Lipomas follow the signal intensity of fat on all MRI sequences, that is, hyperintense on T1, hypointense on fat-saturated sequences, and high signal but hypointense relative to CSF on T2 [ Fig. 8.16 ]. Similarly, they are hyperechoic on fetal/cranial ultrasound and exhibit negative Hounsfield units on CT.

Hindbrain Anomalies
Recent advances in correlation of imaging phenotype with genetics and developmental biology have led to improved understanding and classification of posterior fossa anomalies. A detailed consideration of the hindbrain anomalies is beyond the scope of this text; however, some of the more common anomalies will be considered.
A fundamental principle of understanding hindbrain malformations is that the brainstem and cerebellar development are interrelated, and that abnormalities in one are typically associated with abnormalities in the other. Postformational destructive processes should be suspected when only the cerebellum or brainstem is abnormal.
Malformation of the Cerebellar Vermis
A variety of disorders may be associated with complete or partial absence of the cerebellar vermis. These include entities such as Joubert syndrome, Dandy-Walker spectrum anomalies, and rhombencephalosynapsis.
Joubert syndrome.
Joubert syndrome is a hindbrain malformation with underdevelopment of the cerebellar vermis. Symptoms include hyperpnea, hypotonia, abnormal eye movements, ataxia, and developmental delay. On imaging, the cerebellar vermis is extremely underdeveloped or completely absent, the cerebellar hemispheres are apposed to one another in the midline, and there is enlargement of the superior cerebellar peduncles (“molar tooth sign”) [ Fig. 8.17 ]. Cleft lip and palate may also be present.

Rhombencephalosynapsis.
Rhombencephalosynapsis is a rare disorder in which the cerebellar vermis does not form and there is fusion of the cerebellar hemispheres across the midline producing a diamond-shaped fourth ventricle [ Fig. 8.18 ]. Associated supratentorial anomalies including hypoplasia of the corpus callosum, optic nerves, and agenesis of the posterior lobe of the pituitary gland may be present.

Dandy-Walker spectrum.
Classification of the Dandy-Walker spectrum anomalies continues to be controversial. The term Dandy-Walker spectrum may variably be used to encompass a number of anomalies including the classic Dandy-Walker malformation, the Dandy-Walker variant (less dilatation of the fourth ventricle and less enlargement of the posterior fossa), retrocerebellar arachnoid cysts, Blake pouch cysts (ballooning of the superior medullary velum into the cisterna magna), and mega cisterna magna. The classic Dandy-Walker malformation consists of an enlarged posterior fossa, elevated torcular Herophili, severe hypoplasia or agenesis of the cerebellar vermis, and a dilated, cystic-appearing fourth ventricle [ Fig. 8.19 ]. These anomalies are seen in approximately 1 in 30,000 live births. A number of different gene and modes of inheritance have been implicated.

Other anomalies associated with Dandy-Walker spectrum include hydrocephalus (70–90%), callosal abnormalities (30%), PMG or gray matter heterotopias (5–10%), and occipital encephaloceles in up to 16%. Clinical presentation varies depending on the severity of the abnormality. Eighty percent of patients with classic Dandy-Walker malformation present in the first year of life with symptoms secondary to hydrocephalus.
Abnormalities of Dorsal Induction
Chiari Malformations
Anomalies of the cervicomedullary junction are commonly known as Chiari malformations, after Hans Chiari who described the original three cervicomedullary malformations in 1891.
Chiari I Malformation.
The Chiari I malformation is defined as caudal extension of the cerebellar tonsils below the level of the foramen magnum [ Fig. 8.20 ]. The most common type of Chiari I malformation is related to a congenitally small, bony posterior fossa. Chiari I can also develop in patients with premature closure of the sutures as seen in Crouzon syndrome and Pfeiffer syndrome, and in some infants with ventriculoperitoneal shunts for hydrocephalus. Children with a Chiari I malformation may be asymptomatic or present with irritability (in younger children), headaches, lower cranial nerve palsies, or dissociated extremity anesthesia secondary to syringohydromyelia.

Potentially significant tonsillar descent in Chiari I should be distinguished from milder tonsillar ectopia, which is likely an incidental, clinically silent finding. Although the imaging findings distinguishing “Chiari I” from “tonsillar ectopia” historically focused on the extent of tonsillar ectopia present, clinical symptoms are related to the degree of obstruction of CSF flow at the foramen magnum, and surgical decompression is directed at relieving the obstruction. In Chiari I malformation, the tonsils extend to a variable degree (typically >6 mm), have a “pointed” configuration, and obstruct the flow of CSF at the foramen magnum. Clinically asymptomatic tonsillar ectopia may occur when the tonsils extend into the upper cervical canal, retain a rounded configuration, and do not significantly impede CSF flow. Hydromyelia is present in 20% of patients with a Chiari I malformation. The hydromyelic cavity usually, although not exclusively, involves the cervical spinal cord and extends a variable distance into the thoracic spinal cord. Hydromyelia produces a typical, partially “septated” appearance that serves to differentiate it from syringomyelia [ Fig. 8.21 ]. The demonstration of hydromyelia in the spinal cord should prompt evaluation of the cervicomedullary junction for the Chiari I malformation that will usually be present.

Chiari II Malformation.
The Chiari II malformation is a complex malformation of the hindbrain and skull that occurs in association with an open neural tube defect. This pathogenesis of Chiari II malformations remains controversial, but the anomaly arises in combination with different genetic polymorphisms and in association with environmental factors, including deficient maternal folate intake and abnormalities of folate metabolism. It is relatively common with an incidence of approximately 1 in 1000 live births. Almost all patients with a Chiari II malformation have a myelomeningocele. The hindbrain abnormalities are thought to occur secondary to the failure of closure of the posterior neuropore. This leads to a pressure gradient between the intracranial (higher pressure) and intraspinal (lower pressure spaces) with resultant inferior herniation of the developing structures of the posterior cranial fossa. Most of the hindbrain findings can be thought of as resulting from a normal-sized cerebellum developing in a small posterior fossa with a low-lying tentorium [ Fig. 8.22 ].

Features commonly seen in Chiari II include hydrocephalus, dysplastic tentorium, a small, bony posterior fossa, and a characteristic skull dysplasia known as Lückenschädel skull. Lückenschädel skull results from mesodermal dysplasia, which gives rise to the characteristic scalloped appearance. These dysplastic foci ossify and normalize by 6 months of age.
Other stigmata of Chiari II malformation include inferior displacement and elongation of the brainstem, kinking of the cervicomedullary junction, upward cerebellar herniation, an enlarged massa intermedia, elongated cranial nerves, tectal beaking, and callosal hypogenesis. Approximately 50% of patients have syringohydromyelia. Many patients have small-appearing gyri with shallow sulci in the posterior and medial aspects of the cerebral hemispheres; this pattern is known as stenogyria. Subependymal heterotopia are often present along the trigones of the lateral ventricles.
Chiari II malformations are being more frequently diagnosed antenatally. The classic antenatal sonographic appearance includes the lemon sign (which describes an indentation of the frontal bone) and the banana sign (which describes the appearance of the cerebellum tightly wrapped around the brainstem with obliteration of the cisterna magna). Fetal MRI can add diagnostic information regarding the intracranial abnormalities [ Fig. 8.23 ].

Early neonatal repair of the myelomeningocele and shunting of hydrocephalus has been the standard management of children born with Chiari II. Prenatal surgical repair of the myelomeningocele is now being performed at some centers and has been reported to reduce hydrocephalus, reduce requirement for postnatal ventriculoperitoneal shunt placement, and improve motor outcomes at 30 months. The long-term impact on neurocognitive development of fetal repair of the myelomeningocele has not yet been established.
Chiari III malformation was originally used to describe herniation of posterior fossa contents through a low occipital calvarial defect or high cervical spinal dysraphism. Chiari III malformations are rare.
Anencephaly and Cephalocele
Anencephaly.
Complete failure of early closure of the cephalic end of the neural tube results in anencephaly in which the forebrain, skull, and scalp are absent. It is associated with raised maternal serum alpha-fetoprotein. It is easily detected on antenatal ultrasound [ Fig. 8.24 ]. In the first trimester, neural tissue is still present; however, the normal head contour is absent and crown rump length is less than expected. As the pregnancy continues, neural tube superior to the orbits dissolves and the absent calvarium becomes more apparent. MRI is not required for diagnosis. Anencephaly is invariably lethal within a few days of delivery.

Cephalocele.
A cephalocele refers to herniation of CNS tissue through a defect in the skull. Most cephaloceles are midline in location. They are classified according to their contents and the location of the skull defect. A herniation containing CSF lined by meninges is referred to as a meningocele. A meningoencephalocele contains CSF and brain tissue, and a meningoencephalocystocele contains CSF, brain, and ventricle [ Figs. 8.25 and 8.26 ].

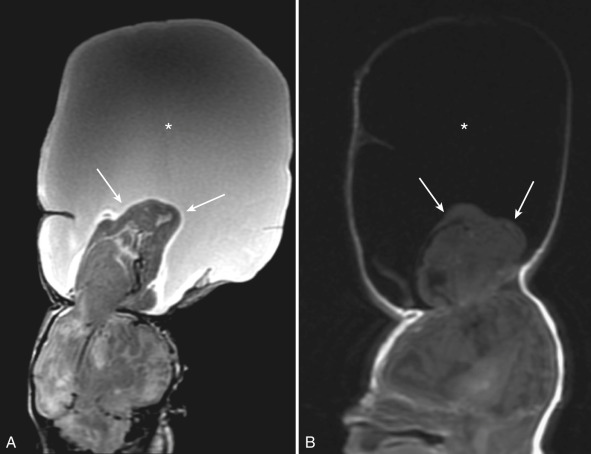
Occipital cephaloceles are the most common. Additional anomalies can be seen in up to 50% of cases including aneuploidy, Dandy-Walker spectrum, and Meckel-Gruber syndrome. Occipital encephaloceles often contain dysplastic occipital or cerebellar tissue, and anomalies of the adjacent dural venous sinuses are common.
Frontal and frontoethmoidal encephaloceles are more common in Asian populations. These may present with hypertelorism or a glabellar mass [ Fig. 8.27 ]. Failure of closure at the foramen cecum may result in a nasofrontal cephalocele, dermoid-epidermoid, or nasal “glioma” (isolated ectopic, dysplastic brain tissue) [ Fig. 8.28 ]. Rare sphenoidal cephaloceles can result in a nasopharyngeal mass.


Cephaloceles can be seen on prenatal ultrasound or fetal MR; they may appear cystic or solid depending on their contents. In postnatal patients, MRI is the best modality for delineating the composition of the contents and intracranial relationships. Thin-section CT with multiplanar reformats can be used for more accurate assessment of the osseous anatomy.
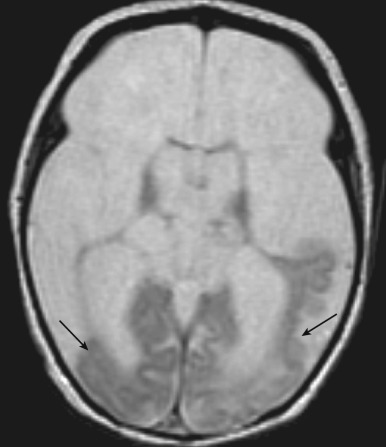
Hydranencephaly
Hydranencephaly is a condition in which most of the cerebral hemispheres are damaged, liquefied, and resorbed in utero, resulting in replacement with a membranous sac containing CSF and the remnants of the cerebral cortex and white matter [ Fig. 8.29 ]. It can occur secondary to a variety of destructive causative factors including bilateral occlusion of the supraclinoid segment of the internal carotid artery (ICA), diffuse hypoxic-ischemic necrosis, necrotizing vasculitis caused by intrauterine infection, and thromboplastic emboli originating in a deceased co-twin.
Craniosynostosis.
Craniosynostosis is the term applied to the premature fusion of a calvarial suture. It can affect the whole or part of a suture and can involve a single or multiple sutures. Failure of growth at the closed sutures causes increased growth along open sutures and resultant craniofacial abnormality [ Table 8.2 ]. Craniosynostosis can be either primary, caused by abnormal suture fusion, or secondary, caused by a failure of brain growth. Isolated single-suture synostosis does not restrict brain development and is usually only of cosmetic importance.
| Suture | Calvarial Configuration | Descriptive Terms |
|---|---|---|
| Sagittal |
| Scaphocephaly |
| Bicoronal |
| Brachycephaly |
| Metopic |
| Trigonocephaly |
| Bilambdoid |
| Turricephaly |
| Unicoronal |
| Plagiocephaly |
| Bicoronal and bilambdoid |
| Cloverleaf or Kleeblattschädel skull |
| All sutures |
| Microcephaly |
Primary craniosynostosis is relatively common with an estimated incidence of 1 in 2000 to 2500 live births. Most cases of craniosynostosis are isolated, but 15% are syndromic and associated with other developmental abnormalities.
Embryologically, the cranial vault is derived from the mesodermal neurocranium. The ossification centers begin to ossify around the 13th gestational week. By the 18th gestational week, the borders of the mineralizing bones have approached each other and begun to form the nonossified sutures. The presence of nonossified sutures and open fontanels at term allows overlap and deformation of the skull bones during parturition. The sutures permit further remodeling during rapid phases of brain and skull growth during the first few years of life.
Sutures normally close from back to front and from lateral to medial. The metopic suture is the exception; it closes from the glabella to the anterior fontanelle and is the first to close (9–11 months). The fontanels normally close by the second year of life. The coronal, lambdoid, and sagittal sutures may remain open until the fourth decade of life.
Sagittal synostosis is the most common isolated synostosis. It is four times more common in males than in females. It clinically manifests as a long, narrow head shape with sagittal ridging, restricted growth the parietotemporal regions, frontal bossing, and occipital protrusion. The long, narrow skull has been described as scaphocephalic, which means “boat-shaped” [ Fig. 8.30 ].

Unicoronal synostosis is the second most common form of craniosynostosis. Two-thirds of affected patients are female. The clinical features include a flattened ipsilateral forehead, a flattened ipsilateral occipital area, the “harlequin eye” deformity (caused by superior displacement of the ipsilateral orbital roof and lesser sphenoid wing), ipsilateral temporal bulging and cheek protrusion, contralateral forehead bossing, and deviation of the nose to the contralateral side [ Fig. 8.31 ].

Bicoronal synostosis causes a short, wide head that can be described as brachycephalic. There is depression of the supraorbital rims with bitemporal and upper forehead bulging. In bicoronal synostosis, the frontosphenoidal and frontozygomatic sutures are typically fused as well.
Bilambdoid synostosis causes a shallow posterior fossa and towering skull known as turricephaly. In a more severe form when bicoronal and bilambdoid synostoses are combined, the skull can adopt a characteristic towering and narrow shape, with bulging temporal areas and shallow orbits known as cloverleaf or Kleeblattschädel skull.
Metopic synostosis, when it occurs before the age of 6 months, is associated with a triangular wedding of the forehead ( trigonocephaly ), hypotelorism, narrow anterior cranial fossa, hypoplasia of the ethmoids, and a metopic ridge. After 6 months of age, premature closure of the metopic suture is associated with minimal deformity.
More than 180 described syndromes are associated with craniosynostosis. The management of syndromic craniosynostosis is complicated by associated skull base, facial, and brain abnormalities. Children with one of the syndromic craniosynostoses are at risk for raised intracranial pressure (ICP), hydrocephalus, optic atrophy, cleft palate, respiratory problems, and hearing disorders [ Table 8.3 ]. The most common of the syndromic causes of craniosynostosis affect the coronal sutures. Bicoronal synostosis may be associated with hydrocephalus, most often caused by jugular foraminal stenosis and venous hypertension.
| Syndrome | Gene Affected | Clinical Findings |
|---|---|---|
| Apert syndrome | FGFR2 | Bicoronal craniosynostosis, midface hypoplasia, hypertelorism, exorbitism, complex syndactyly |
| Crouzon syndrome | FGFR2 | Bicoronal craniosynostosis, midface hypoplasia, hypertelorism, exorbitism |
| Pfeiffer syndrome | FGFR1 or FGFR2 | Multiple craniosynostoses, midface hypoplasia, hypertelorism, severe exorbitism, broad thumbs, broad hallux, soft tissue syndactyly |
| Muenke syndrome | FGFR3 | Coronal synostosis, thimblelike middle phalanges, carpal/tarsal coalition, sensorineural hearing loss |
Apert syndrome (also known as type I acrocephalosyndactyly) is an autosomal dominant condition that results from mutations of the FGFR2 gene. It is characterized by bicoronal craniosynostosis, midface hypoplasia, hypertelorism, exorbitism, and severe complex syndactyly of fingers and toes [ Fig. 8.32 ].

Crouzon syndrome is also due to mutations of the FGFR2 gene. Crouzon syndrome has similar calvarial deformities, facial anomalies, and exorbitism but is not associated with hand and foot abnormalities.
Pfeiffer syndrome (also known as type V acrocephalosyndactyly) can occur because of mutations in FGFR1 or FGFR2. It is characterized by craniosynostosis affecting multiple sutures, midfacial hypoplasia, severe exorbitism, and hypertelorism. The clinical findings in the periphery include broad and medially deviated great toes, broad thumbs, and soft-tissue syndactyly.
Muenke syndrome is caused by a mutation in the FGFR3 gene and is strongly associated with coronal synostosis. The hands and feet are often affected but with mild abnormalities such as thimble-like middle phalanges, carpal or tarsal coalition, and coned epiphyses that may not be clinically significant. All patients with Muenke syndrome should be tested for sensorineural hearing loss, but only a few are actually affected.
Imaging plays an important role in confirming the diagnosis and management of craniosynostosis, evaluating the extent of involvement, which is difficult with clinical examination alone. CT is the primary imaging modality used to evaluate the sutures. The closed suture is demonstrated by bone bridging across the suture, loss of architecture, or ridging. A very-low-dose technique can be used when the primary concern is osseous anatomy, and 3D reconstructions can depict the abnormalities exquisitely and are critical for operative planning.
Imaging can also help differentiate lambdoid craniosynostosis from positional plagiocephaly, which is an abnormal skull shape caused by infant positioning and not by premature fusion of the sutures [ Fig. 8.33 ]. There has been an increase in occipital plagiocephaly over the past two decades because of “back to back” campaigns encouraging that infants sleep in the supine position to reduce the risk for sudden infant death syndrome. In positional plagiocephaly, in addition to the lambdoid sutures being open, the ear on the flattened side is “pushed” forward; but in lambdoid synostosis, the ipsilateral ear is “pulled” posteriorly toward the fused suture.

In complex cases, dedicated magnetic resonance venography (MRV) or computed tomographic venography (CTV) imaging may be performed preoperatively to assess for associated dural venous sinus anomalies. In bicoronal synostosis with jugular foraminal stenosis, enlarged transosseous emissary veins (sinus pericranii) may be present. MRI may be required if there is a need to assess for underlying parenchymal abnormalities inadequately imaged using the low-dose CT technique.
Surgical intervention is unique to each type of synostosis. Surgery may be undertaken to correct cosmetic deformity in single-suture synostosis or may be required to permit normal brain growth or prevent complications such as raised ICP in multisutural synostosis.
Neurocutaneous Syndromes
The phakomatoses are a collection of hereditary disorders that are characterized by hamartomas and other congenital malformations involving structures arising from the ectoderm such as the nervous system, skin, retina and globe. The phakomatoses are also referred to as neurocutaneous disorders because of their involvement of both skin and neural tissues. Some of the more common phakomatoses include neurofibromatosis, tuberous sclerosis complex (TSC), Sturge-Weber disease, von Hippel-Lindau disease (VHL), and ataxia-telangiectasia.
Neurofibromatosis
Neurofibromatosis has multiple subtypes. The two most common are types I and II.
Neurofibromatosis Type 1
Neurofibromatosis type 1 (NF-1) is one of the most common autosomal dominant conditions that affect the CNS. It was first described by von Recklinghausen in 1885. Its incidence is estimated at 1 in 2000 to 3000 live births. The NF1 gene has been mapped to chromosome 17q11.2 and produces a cytoplasmic protein called neurofibromin. In the CNS, this protein is mainly expressed in neurons, Schwann cells, oligodendrocytes, and astrocytes.
The phenotypic expression of NF1 is variable. A diagnosis requires two or more of the following clinical criteria: (1) six or more café-au-lait spots, (2) two or more neurofibromas or one or more plexiform neurofibromas, (3) axillary freckling, (4) optic pathway glioma (OPG), (5) two or more Lisch nodules of the iris, (6) distinctive bone dysplasia or thinning of long-bone cortex, and (7) first-degree relative with NF-1 [ Box 8.1 ].
Diagnosis requires two or more of the following criteria:
- 1.
Six or more café-au-lait spots
- 2.
Two or more neurofibromas or one or more plexiform neurofibromas
- 3.
Axillary freckling
- 4.
OPG
- 5.
Two or more Lisch nodules of the iris
- 6.
Distinctive bone dysplasia or thinning of long-bone cortex
- 7.
First-degree relative with NF-1
Café-au-lait spots are the earliest manifestation of NF-1 to develop and are present in more than 95% of patients. Axillary freckling develops in approximately two-thirds of patients. Cutaneous neurofibromas and Lisch nodules increase in number throughout life. Cognitive impairment occurs in up to 50% of patients.
Ninety percent of children with NF-1 have characteristic multiple T2/FLAIR hyperintense lesions that can be found in the brainstem, cerebellar white matter, basal ganglia, thalamus, internal capsule, and occasionally the corona radiata [ Fig. 8.34 ]. These were previously referred to as “unidentified bright objects” or “NF-1 spots.” It is now known that these foci represent focal areas of myelin vacuolization. These lesions regress late in the first decade of life and are rarely seen after the age of 20 years. Features that help to distinguish these lesions from astrocytomas include their characteristic location, isointensity or slight hyperintensity on T1-weighted sequences, and lack of enhancement. Although NF-1 spots do not have mass effect, the brain may be slightly malformed locally, especially in the brainstem and middle cerebellar peduncle. This deformity and signal abnormality may lead to the erroneous diagnosis of brainstem glioma if other clinical or imaging stigmata of NF-1 are not recognized.

The most important primary CNS abnormality seen in NF-1 is OPG, which occurs in 15% to 20% of patients. OPGs tend to occur before the age of 7 years and can involve a single optic nerve, both nerves, the chiasm, and posterior optic pathways [ Fig. 8.35 ]. OPG in patients with NF-1 tends to have a more indolent course than sporadic OPG. Some optic pathway tumors in NF-1 even spontaneously involute with time. Most OPGs seen in NF-1 are histologically low-grade pilocytic astrocytomas. The optic nerves are often tortuous in NF-1, even in the absence of OPG. Optic pathway tumors may be either nonenhancing or may enhance avidly. Astrocytomas in locations other than the optic pathway are also more common in NF-1 than in the general population. Apart from OPGs, cranial nerve tumors are rare in NF-1 in contrast with neurofibromatosis type 2 (NF-2).

Macrocephaly and enlargement of the corpus callosum can also be seen in NF-1. Up to 6% of patients with NF-1 have cerebral vascular dysplasia. This most commonly consists of intimal and smooth muscle proliferation causing multifocal stenosis and occlusion of the internal carotid terminus and proximal middle cerebral artery (MCA) and anterior cerebral artery (ACA). This results in a moyamoya vasculopathy pattern with proximal steno-occlusive change and prominent lenticulostriate and thalamoperforate or collaterals in many such patients [ Fig. 8.36 ]. The vasculopathy may occur even without a history of radiation for suprasellar glioma in children with NF-1.

Calvarial and orbital abnormalities are seen in NF-1, with the classic lesion being sphenoid wing dysplasia [ Fig. 8.37 ]. Craniofacial neurofibromas and plexiform neurofibromas can secondarily cause intracranial complications. Neurofibromas are characterized on MRI by central T2 hypointensity giving rise to the “target sign.” Plexiform neurofibromas are larger, more extensive, and tend to be more heterogeneous in signal intensity. Spinal manifestations of NF-1 include scoliosis, intramedullary tumors, dural ectasia, lateral meningocele, and nerve root neurofibromas.

Neurofibromatosis Type 2
NF-2 is a separate and distinct disease from NF-1. The NF-2 gene maps to chromosome 22q12 and produces a protein called merlin or schwannomin. NF-2 is autosomal dominant but much less prevalent than NF-1, with an estimated incidence of 1 in 50,000. Fifty percent of cases are sporadic. Manifestations of NF-2 are extremely uncommon in children. The major clinical feature of NF-2 is the presence of bilateral vestibular (CN VIII) schwannomas in nearly all patients. Other manifestations can be remembered using the mnemonic MISME ( m ultiple i nherited s chwannomas, m eningiomas and e pendymomas) [ Fig. 8.38 ]. Simplistically, NF-2 can be thought of as involving tissues that surround neural structures, whereas NF-1 typically affects the neural structures directly.

Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


