Liver and Biliary Tree
Anatomy and Embryology
The liver, gallbladder, and biliary tree originate from endodermal cells that form a diverticulum arising from the duodenal region of the primitive embryonic gut between 4 and 10 weeks’ gestation. The larger cranial division (pars hepatica) gives rise to the liver, whereas the smaller, caudal portion (pars cystica) develops into the gallbladder and cystic duct. The intrahepatic and extrahepatic components of the biliary tree develop independently and unite by 12 weeks’ gestation. At birth, the liver represents about 5% of total body weight (200 g). Bile secretion commences between 12 and 16 weeks’ gestation. Hematopoiesis normally occurs only in the fetal liver and ceases by the age of 6 weeks in healthy infants.
Although the liver is usually located in the right upper quadrant, it may be midline and more symmetric in patients with heterotaxy syndromes [ Fig. 5.1 ]. This is seen in approximately 80% of patients with asplenia and about 50% of patients with polysplenia.
The portal venous system supplies between 70% and 80% of the blood supply to the liver. The portal venous branches, hepatic arterial branches, and biliary radicles run parallel to one another in the center of hepatic segments, the so-called portal triad arrangement. Accessory lobes of the liver are rare. Caudal elongation of the right lobe of the liver, known as a Riedel lobe, is a normal variant.
Developmental Anomalies
Biliary Atresia
Jaundice in an infant older than 2 weeks should raise suspicion for underlying liver disease. The clinical differential diagnosis of neonatal jaundice is broad. It may be related to sepsis, hemolysis, infection (cytomegalovirus, hepatitis A and B, rubella), and metabolic abnormalities (α 1 -antitrypsin deficiency, cystic fibrosis [CF]). When these causes of jaundice have been excluded, neonatal hepatitis and biliary atresia account for more than two-thirds of the remaining cases of conjugated hyperbilirubinemia in the neonate. It is postulated that neonatal hepatitis and biliary atresia are part of the same clinical spectrum of cholestatic jaundice and hepatomegaly.
Biliary atresia is relatively rare in the United States, with an incidence of 1/10,000 to 20,000 infants. Progressive ductal inflammation leads to fibrosis and obliteration of the extrahepatic bile ducts, with occasional involvement of the intrahepatic bile ducts. Disruption of the biliary system results in chronic cholestasis, which eventually leads to liver fibrosis and biliary cirrhosis. Obliteration of the bile ducts may be present at the time of birth or may occur shortly after birth. Although there are many hypotheses regarding the etiology of this disease, the exact cause remains unknown.
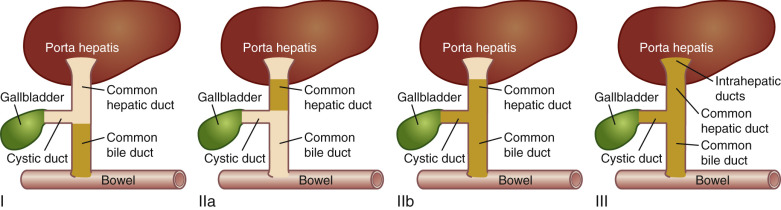
The Kasai system is most commonly used to classify biliary atresia based on location of disease involvement [ Fig. 5.2 ]. The diagnosis of biliary atresia requires definitive imaging of the biliary ductal system. Intraoperative cholangiogram is considered the gold standard for evaluation of the biliary tree, and liver biopsy is needed for microscopic evaluation to determine involvement of the intrahepatic bile ducts. These are invasive procedures and not without significant risk in the infant population. Thus evaluation often begins with ultrasound and hepatobiliary scintigraphy to determine which patients should go on to more definitive diagnostic procedures.
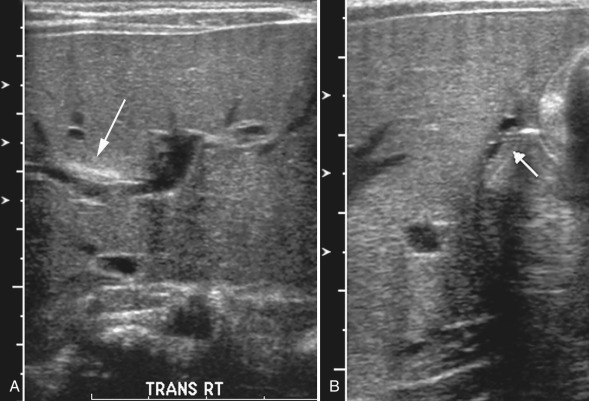
Findings on ultrasound include the “triangular cord sign,” an abnormal triangular or tubular echogenic band of fibrous tissue in the porta hepatis representing the obliterated common hepatic duct [ Fig. 5.3 ]. Because this structure lies immediately adjacent to the portal vessels, it can sometimes appear as a relative thickening of the anterior wall of the right portal vein. On a longitudinal ultrasound image, an anterior wall thickness of greater than 4 mm can be used to define the triangular cord sign.
In 25% of cases of biliary atresia, the gallbladder is absent. If the gallbladder is identified, it is often abnormal. Ultrasound evaluation of the gallbladder before and after feeding can be helpful because the abnormal gallbladder in biliary atresia will not typically demonstrate the expected change in size between the fasting and recently fed states. Ultrasound can also help to identify alternative causes of cholestasis, such as a choledochal cyst or other mass causing biliary obstruction.
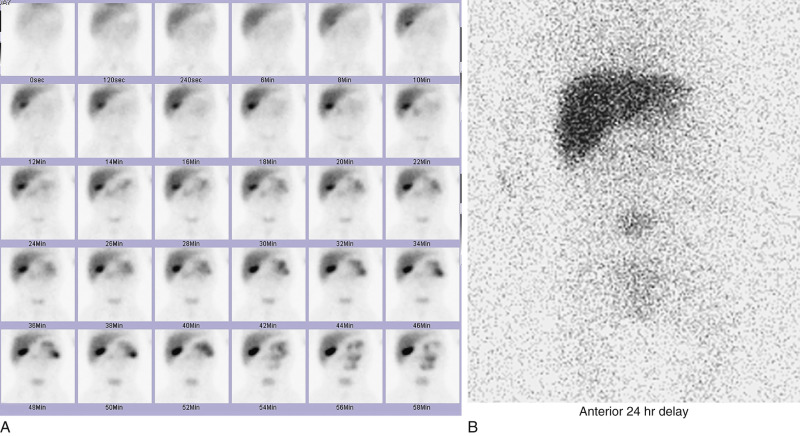
Hepatobiliary scintigraphy with 99m Tc-iminodiacetic acid derivatives is often the second diagnostic imaging study used to evaluate biliary atresia. In the normal liver, radiotracer is taken up and excreted by hepatocytes into the biliary tree within 10 to 15 minutes of venous injection. In biliary atresia, the tracer cannot be excreted and remains in the liver parenchyma. A lack of tracer excretion into the small bowel by 24 hours after tracer injection is suggestive of biliary atresia [ Fig. 5.4 ]. This finding is notably nonspecific and can be seen in any disorder that results in cholestasis. Poor hepatocellular function, as may be seen in neonatal hepatitis, can also result in delayed biliary excretion. Thus an infant undergoing a hepatobiliary scan is typically pretreated with phenobarbital for 5 days before the examination. This enhances hepatocellular function and decreases the chance of a false-positive result. If an infant has not been pretreated with phenobarbital and there is no tracer excretion into the small bowel at 24 hours, an infusion of ursodiol can be given, with repeat imaging at 48 and 72 hours.
Treatment of biliary atresia is surgical, with the best outcomes seen when surgery is performed before 40 to 60 days of life. The surgery of choice is the Kasai procedure. The abnormal extrahepatic biliary tree is mobilized from the liver, and the cut liver surface is directly anastomosed to a Roux-en-Y loop of small bowel. This allows direct drainage of bile from the liver. After 3 months of age, intrahepatic bile duct involvement and resultant hepatic fibrosis are often severe enough to render the Kasai procedure ineffective. In these patients, and in those who undergo a Kasai procedure with no significant improvement in cholestasis, liver transplant is the only treatment option.
Choledochal Cyst
A choledochal cyst is a localized cystic dilatation of the biliary ductal system. It is postulated that this develops when there is an anomalous junction of the common bile duct and the pancreatic duct, forming a long common channel and allowing for reflux of pancreatic enzymes into the biliary tree. This leads to inflammation and focal duct stenosis, with resultant proximal dilation or outpouching of the bile duct. Choledochal cysts occur four times more commonly in females than in males, and they have a high prevalence among individuals from East Asia.
A choledochal cyst is a rare cause of neonatal cholestatic jaundice; however, when identified in this age group, concomitant biliary atresia should always be ruled out.
In older patients, clinical manifestations of a choledochal cyst include abdominal pain, jaundice, and a palpable right upper quadrant “mass.” This complete constellation of clinical findings is notably seen in less than 60% of patients. Laboratory workup reveals a direct hyperbilirubinemia. Most patients present before the age of 10 years.
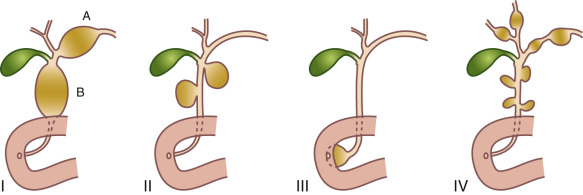
Four types of congenital biliary cysts have been described [ Fig. 5.5 ]. The most common is type 1 (80%–90% of cases), which is confined to the extrahepatic bile ducts. Type 2 is a diverticulum arising from the common bile duct (2% of cases). Type 3 occurs just proximal to the ampulla of Vater in the distal intraduodenal wall segment of the common bile duct, with extension into the duodenal lumen. Type 4 consists of multiple regions of cystic dilatation of the extrahepatic or intrahepatic biliary tree.
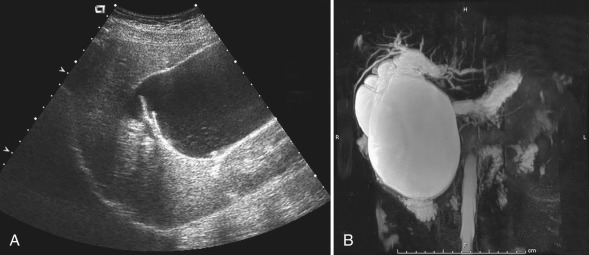
Ultrasound is a useful imaging modality for the evaluation of choledochal cysts. Cysts appear as anechoic structures in the region of the porta hepatis or under the liver, separate from the gallbladder and in direct communication with the biliary tree [ Fig. 5.6 ]. Enlargement of the proximal common bile duct or dilation of the intrahepatic ducts can be seen. If a choledochal cyst is suspected, the entire biliary tree should be closely interrogated to identify the extent of disease. With significant cystic dilation of the common bile duct, biliary lithiasis can be seen secondary to bile stasis. Magnetic resonance imaging (MRI) and computed tomography (CT) show findings similar to ultrasound, and can also be used to better define any associated intrahepatic disease. Hepatobiliary scanning may be useful to confirm communication between the cyst and the hepatobiliary tree. However, it yields little additional anatomic information.
Caroli Disease
There are two forms of Caroli disease; both are rare inherited disorders. The most common form is an autosomal dominant disorder characterized by cystic dilatation of the intrahepatic biliary tree with no evidence of biliary obstruction. Patients may present from infancy through young adulthood with clinical symptoms of fever, intermittent abdominal pain, and hepatomegaly. This form of Caroli disease can be complicated by cholangitis, stone formation, and rarely, cholangiocarcinoma.
The second form of the disease is called Caroli syndrome . This is a more complex, autosomal recessive condition associated with congenital hepatic fibrosis and autosomal recessive polycystic kidney disease. The genetic basis for the hepatic and renal abnormalities is a series of mutations in the PKHD1 gene that result in malformed biliary ducts and renal collecting tubules.
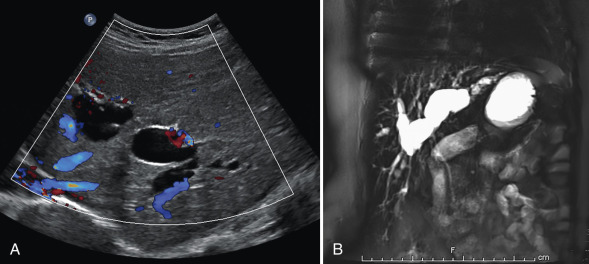
Caroli disease has a distinct pattern on cross-sectional imaging, with fusiform or saccular dilation of the intrahepatic bile ducts [ Fig. 5.7 ]. Also seen is the classic “central dot sign,” which reflects a portal vein radical surrounded by a dilated bile duct. These findings can be seen with sonography, MRI, and CT. In patients with Caroli syndrome, imaging findings consist of hepatomegaly, dilatation of the biliary radicles, and fibrous replacement of liver parenchyma. Hepatic fibrosis progressively leads to symptomatic portal hypertension.
Antibiotics can be used to treat intercurrent infections, and ursodeoxycholic acid can be administered for stone formation. Endoscopic stent placement and liver transplantation have been used successfully in selected patients.
Acquired Conditions
Cholelithiasis and Choledocholithiasis
Gallstones are a common finding in children. In infants, hepatic immaturity is associated with decreased secretion of bile salts, and immature biliary conjugation pathways contribute to the formation of pigmented stones and bile sludge. Predisposing factors include the use of diuretics (furosemide) in premature infants, the administration of total parenteral nutrition, extended fasting, and an altered enterohepatic circulation, as may be seen in infants with short bowel syndrome. In these clinical settings, stones often resolve spontaneously.
Cholelithiasis is idiopathic in approximately 40% of older children. Hemolytic diseases such as sickle cell disease and thalassemia are responsible for an additional 30% of cases. Other conditions including inflammatory bowel disease affecting the ileum, CF, obesity, and short gut syndrome (after bowel surgery) have also been associated with gallstone formation. With these entities, an interrupted enterohepatic circulation is contributory.
It is important to remember that up to 50% of pediatric patients will be asymptomatic with gallstones. Another large percentage of patients will present with nonspecific, intermittent abdominal pain. The most common complications of gallstones in pediatric patients are acute calculous cholecystitis and acute pancreatitis in the setting of cystic duct or pancreatic duct obstruction.
Although conventional abdominal radiographs may demonstrate up to 50% of gallstones, ultrasound is considered the imaging modality of choice for evaluation, with an accuracy of more than 95% for the detection of cholelithiasis. Luminal debris/sludge, bile concretions, and calculi are all well demonstrated by ultrasound. Dilatation of the common duct is often the only indirect sign of downstream calculi because direct stone visualization may be difficult in the setting of overlying bowel gas.
Even though the appearance of most gallstones on CT is identical to that seen on radiographs, some stones will be isodense to background bile in the gallbladder, and thus can be missed on CT.
Gallstones are seen on MRI as T2 hypointense foci against a background of T2 bright bile in the gallbladder. Stones can also be evaluated with magnetic resonance cholangiopancreatography (MRCP), which will demonstrate filling void(s) in the gallbladder or biliary tree.
In asymptomatic patients, treatment is typically expectant with periodic ultrasound surveillance. Patients who are symptomatic from gallstones should have them removed. Endoscopic retrograde cholangiopancreatography (ERCP) is often the treatment of choice in symptomatic patients without predisposing underlying disease. If ERCP is unsuccessful, laparoscopic cholecystectomy is performed.
Biliary Sludge
Noncalcified biliary debris (sludge) is a common, transient phenomenon resulting from biliary stasis. Predisposing conditions include biliary obstruction, extended fasting, parenteral nutrition, and hemolysis. Although usually asymptomatic, the presence of biliary sludge may be associated with a dramatic but transient rise in conjugated bilirubin. On sonographic evaluation, sludge is moderately echogenic and usually lacks acoustic shadowing. It may be associated with a fluid–debris level or may coalesce into focal echogenic “masses” (tumefactive sludge) [ Fig. 5.8 ]. Sludge is typically mobile with changes in patient position.
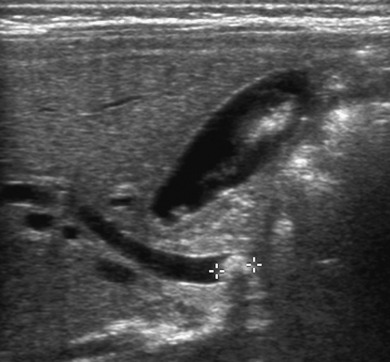
Biliary sludge does not require specific therapy in most situations. However, frequent sonographic monitoring is useful to document resolution or progression to calculus formation.
Cholecystitis
Acute cholecystitis occurs far less commonly in children than in the adult population. However, mortality rates in young children approach 30% because of associated serious concomitant illness and a high risk for ischemia and perforation. Cholecystitis may be classified as calculous or acalculous based on the presence or absence of obstructing calculi in the bladder neck or cystic duct. The common pathway in both conditions involves distention of the gallbladder, with increased intraluminal pressure and bile stasis. Bile stasis results in chemical injury to the gallbladder mucosa, with progressive edema, ischemia, and necrosis of the gallbladder wall. Perforation may occur in severe cases. Acalculous cholecystitis constitutes more than 50% of cases in infants and young children and has been associated with severe gram-negative bacterial infections, cytomegalovirus, and Epstein-Barr virus [ Fig. 5.9 ].
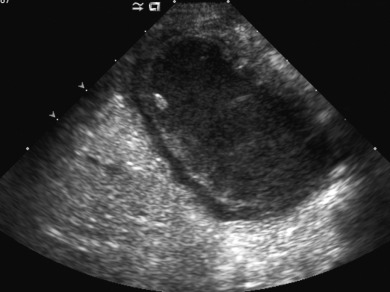
Sonography is the imaging modality of choice to diagnose acute cholecystitis. Ultrasound findings in pediatric patients are similar to those seen in adults. An obstructing biliary stone often can be identified in the gallbladder neck, cystic duct, or common bile duct. A positive sonographic Murphy sign may be helpful when present, although this finding is not always reliable in the pediatric population. Gallbladder wall thickening (>3 mm) and pericholecystic fluid are important secondary findings. An additional secondary finding is edema involving the adjacent liver parenchyma.
Gallbladder wall thickening should be approached with caution because this finding can be seen in multiple pathologic states. The differential diagnosis of gallbladder wall thickening is broad and includes pseudothickening in the postprandial state, acute or chronic cholecystitis, reactive change secondary to acute hepatitis, edema in the setting of congestive heart failure, and edema related to third-spacing in hypoalbuminemia.
Findings of acute cholecystitis on CT and MRI are similar to those seen with ultrasound. MRI may be more sensitive for the detection of inflammation within the gallbladder wall and in the liver and mesentery surrounding the gallbladder. Evaluation of the biliary tree with MRCP can demonstrate an impacted stone in the gallbladder neck or cystic duct. Although frequently used in the past, CT is not currently recommended for this clinical indication in pediatric patients because of the associated radiation exposure.
Hepatobiliary scanning is not commonly used in the pediatric population for the diagnosis of biliary obstruction. It is typically reserved for cases with high clinical suspicion and equivocal findings on ultrasound. The most common positive scintigraphic finding in pediatric acute cholecystitis is lack of visualization of the gallbladder by 40 minutes after tracer administration.
Medical treatment consists of supportive measures, intravenous antibiotics, and occasional percutaneous cholecystotomy before definitive surgery.
Hydrops of the Gallbladder
Gallbladder hydrops is characterized by marked distension of the gallbladder without evidence of secondary inflammation. It occurs in the setting of transient, self-limiting acalculous obstruction of bile flow. On imaging, the hydropic gallbladder is often longer than the adjacent right kidney but maintains normal wall thickness with no appreciable hyperemia or pericholecystic fluid. Both gallbladder hydrops and acute acalculous cholecystitis may occur in Kawasaki disease, presumably because of direct vascular compromise to the gallbladder.
Cholangitis
Ascending cholangitis may be caused by a number of conditions, including calculi, parasitic infestation, immunodeficiency syndromes, and biliary obstruction caused by neoplasms. It may also occur as a complication of surgical (Kasai) repair of biliary atresia. Sclerosing cholangitis is typically an autoimmune disease. More than 80% of patients with primary sclerosing cholangitis (PSC) have concomitant inflammatory bowel disease, and about one-third of patients have associated autoimmune hepatitis. Other diseases associated with sclerosing cholangitis in children include Langerhans cell histiocytosis and CF. Patients typically present in the second decade of life with jaundice, hepatomegaly, and abdominal pain.
Dilatation and irregular wall thickening of the common bile duct and large intrahepatic ducts are seen on ultrasound, CT, and MRI. At sonography, hyperechoic portal triads, a thickened gallbladder wall, and calculi are also demonstrated in patients with PSC. Abnormal contrast enhancement of the biliary duct walls may be seen on contrast-enhanced CT. In many pediatric institutions, MRI with MRCP is the preferred imaging modality in patients with suspected PSC. This technique well demonstrates focal areas of hepatic parenchymal signal abnormality and a beaded appearance of the common bile duct and large intrahepatic ducts [ Fig. 5.10 ]. In older children, MRCP has a reported sensitivity greater than 85% and a specificity greater than 90% for the detection of PSC.
Treatment with ursodeoxycholic acid alone or in combination with immunosuppressive therapy may temporarily contain disease activity. However, up to one-third of patients with PSC will eventually go on to require liver transplantation.
Hepatic Parenchymal Disorders
Hepatic Infections
Viral Hepatitis.
Viral hepatitis is the most common diffuse infection of the liver in otherwise healthy children. In the newborn, cytomegalovirus and hepatitis B virus are the most common causative agents associated with chronic inflammatory change, fibrosis, and eventual cirrhosis. In older children, viral hepatitis is most commonly caused by the hepatitis A, B, and C viruses, although cytomegalovirus, herpes simplex virus, varicella zoster virus, and Epstein-Barr virus can all result in hepatic inflammation.
The diagnosis of viral hepatitis is made clinically and on the basis of laboratory data. The clinical presentation of viral hepatitis varies with the pathogen. Hepatitis A presents with a flu-like illness and fever, and is sometimes associated with jaundice. In children, the disease is typically self-limited and may be clinically inapparent. Both hepatitis B and C cause acute disease, which may progress to chronic hepatitis and possibly the development of cirrhosis and hepatocellular carcinoma (HCC).
If imaging is performed during the early stages of illness, the liver is usually normal in appearance. As the disease progresses, there may be hepatomegaly, and the liver parenchyma may appear somewhat echogenic and heterogeneous on ultrasound. In severe acute hepatitis, there is diffusely decreased parenchymal echogenicity with relative increased echogenicity of the portal triads, resulting in the classic “starry-sky” appearance. Other findings on ultrasound include enlargement of lymph nodes in the porta hepatis and reactive inflammatory change involving the gallbladder, including gallbladder wall thickening and pericholecystic fluid. CT may show mild hepatomegaly, heterogeneous liver parenchyma, decreased periportal attenuation, and gallbladder wall thickening. On MRI, there may be periportal hyperintensity on T2-weighted sequences. Unfortunately, the imaging findings of acute hepatitis are nonspecific and require appropriate clinical correlation.
In some children, viral hepatitis may result in fulminant hepatic failure, characterized by severe impairment of hepatic function with necrosis of hepatocytes in the absence of preexisting liver disease. Necrotic areas contract in size and may have internal hemorrhage or inflammatory cell infiltration.
Pyogenic Abscess.
Pyogenic abscesses are typically polymicrobial, with the most common agents being gram-negative aerobic and anaerobic organisms such as Escherichia coli and Klebsiella and gram-positive organisms such as Staphylococcus aureus and Streptococcus pneumoniae.
The most common route of entry is via the portal venous system in the setting of an intraabdominal infection such as appendicitis or colitis. Abscesses are typically solitary and most commonly seen in the right hepatic lobe. In the very early stages of infection, a cluster of microabscesses may be seen that eventually coalesce into a larger confluent pyogenic abscess cavity. Bacteria may also enter the liver through the hepatic arterial system in the setting of sepsis, resulting in multifocal peripheral abscesses. Biliary entry is also possible after ERCP or biliary surgery. Resultant abscesses are more centrally located or along the path of the intrahepatic biliary tree. Superinfection of necrotic tissue can occur after hepatic trauma.
On cross-sectional imaging, the appearance of pyogenic abscesses may range from a single, well-defined, homogeneous lesion to poorly defined, heterogeneous regions of distorted hepatic parenchyma [ Fig. 5.11 ]. Internal septations and debris can be seen when abscesses are large. There is often increased surrounding vascularity on color Doppler sonography and peripheral enhancement on CT and MRI. All abscesses lack blood flow centrally. Internal gas can be seen in infection with gas-forming organisms. A thin surrounding rim of hepatic edema may be noted.

Treatment of a pyogenic hepatic abscess varies based on size. Smaller abscesses (<3 cm) are typically treated with intravenous antibiotic therapy. Larger collections usually warrant percutaneous aspiration and catheter drainage. Surgical drainage is performed when catheter drainage fails or treatment of an underlying cause of the abscess is required.
Fungal Infection.
Hepatic fungal infection occurs frequently in the setting of disseminated fungal disease in immunocompromised patients. In pediatrics, this includes patients on chemotherapy and those who are status post bone marrow or solid organ transplant. The most common causative organism is Candida albicans , which typically presents with microabscesses involving multiple organs including the liver, spleen, and occasionally the kidneys. Other less common causative organisms include Aspergillus , Coccidioides , Cryptococcus , and Histoplasma . Colonization of the gastrointestinal (GI) tract is the preliminary path of dissemination of Candida , with the presence of mucositis in immunocompromised patients facilitating spread of the organism.
Notably, the diagnosis of hepatic fungal disease is limited on all imaging modalities when the patient is neutropenic and unable to mount a sufficient immune response. In these patients, it is important to consider repeat imaging after recovery of the neutrophil count, especially if clinical suspicion is high and the patient is not responding to conventional antibiotic therapy.
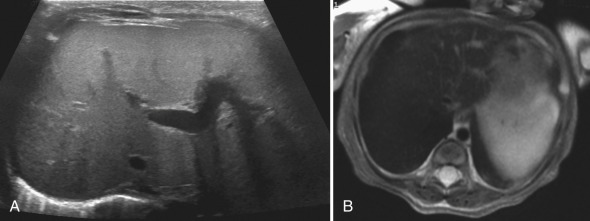
Four sonographic patterns of hepatic candidiasis have been described, representing the different stages of hepatic infection. The “wheel-within-a-wheel” and “bull’s-eye” or “target” appearances are commonly seen during the active phase of infection. The most common pattern seen later in the course of disease is diffuse, tiny (<4 mm), uniformly hypoechoic foci throughout the liver, reflecting the formation of small microabscesses [ Fig. 5.12 ]. Punctate echogenic foci generally indicate healing or healed fungal infection.

Findings on noncontrast CT include multiple foci of low attenuation of varying size, generally between 2 and 20 mm. Microabscesses usually enhance centrally, although peripheral enhancement may occur.
As with ultrasound, the imaging appearance of hepatic fungal disease on MRI varies with the phase of infection and stage of treatment [ Fig. 5.12 ]. Several authors suggest that MRI is superior to CT for the identification of hepatosplenic fungal disease, with a reported sensitivity of 100% and specificity of 96%. Thus many institutions now use limited MRI protocols consisting of fast-acquisition T2-weighted sequences rather than CT for rapid imaging assessment for fungal disease in pediatric patients. More extensive MRI protocols are used to follow disease change after the implementation of treatment.
Parasitic Infections.
Although rare in developed countries, parasitic disease is estimated to affect almost 1 billion people worldwide. The three most common infections—ascariasis, echinococcosis, and amebiasis—are endemic in many parts of the world.
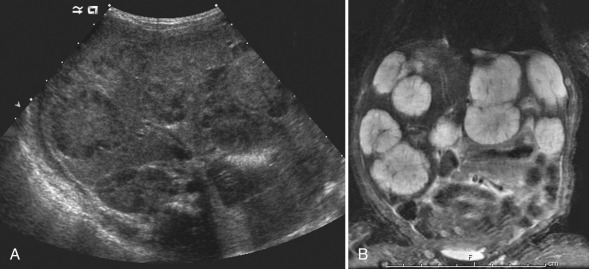
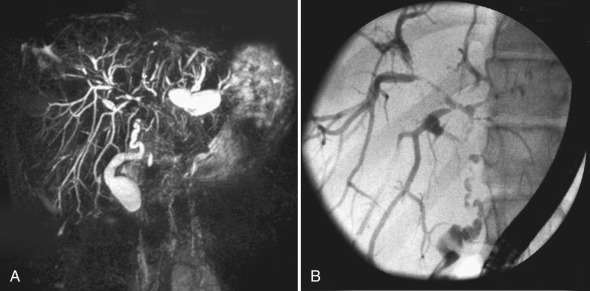
Ascariasis is a parasitic infection that is prevalent in tropical and subtropical regions of the world. It occurs when the parasite eggs are inadvertently ingested. Eggs develop into larvae in the bowel and subsequently develop into long, thin, pencil-shaped worms that penetrate through the intestinal wall, enter the portal venous system, and eventually travel to the liver. Clinical symptoms depend on the stage of infection. Biliary obstruction can occur, resulting in dilation of the intrahepatic bile ducts on ultrasound. Occasionally, worms can be identified within the bile ducts or the portal vasculature [ Fig. 5.13 ].

Echinococcal, or hydatid disease, is somewhat more common. It is endemic in regions of South America, the Middle East, and the Mediterranean. After ingestion of eggs from contaminated soil or water, the protective covering of echinococcal eggs is broken down by gastric acid, and larvae pass through the mucosa into the portal venous system. Larvae then become lodged in the liver and slowly grow into hydatid cysts. Daughter cysts, a pathognomonic finding, form from brood capsules along the periphery of an endocyst [ Fig. 5.14 ]. As they enlarge, echinococcal cysts are prone to rupture. In a “contained rupture,” the endocyst membrane pulls away from the pericyst, resulting in a “floating” internal membrane. If rupture is not contained, cystic contents can spill into the peritoneal cavity and result in seeding of the peritoneum.

Definitive treatment of an echinococcal cyst requires removal of the entire cyst and its contents. Primary surgical removal is often not attempted because of concern for cyst rupture and peritoneal spillage. Initial treatment typically consists of percutaneous drainage in conjunction with antiparasitic therapy with albendazole. This is subsequently followed by complete surgical excision.
An amebic abscess may have a variable imaging appearance. Features are often similar to those of a pyogenic abscess. Both pyogenic and amebic abscesses are more common in the right hepatic lobe and are often peripheral in location near the liver capsule. Treatment of an amebic abscess is medical. Metronidazole is typically administered, with a rapid response most often observed within 24 to 72 hours.
Hepatic Steatosis
Hepatic steatosis is caused by excessive accumulation of triglycerides within hepatocytes. It is the most common cause of chronic liver disease in pediatric patients. The pathologic diagnosis of hepatic steatosis is made when more than 5% of the total liver weight is replaced by fat. Adolescents are affected more often than younger children, with a higher prevalence in boys than girls. Obesity and associated insulin resistance are the most common risk factors for hepatic steatosis. The overall incidence of disease is increasing in the United States with the increase in pediatric obesity. Fatty infiltration of the liver may be focal or diffuse. When focal, fatty deposition classically occurs along the falciform ligament and the gallbladder fossa. Areas of focal fat deposition demonstrate well-defined margins.
Metabolic Diseases
Several hereditary metabolic diseases may cause liver damage with liver failure or cirrhosis, with or without associated injury to other tissues. These include hemochromatosis, tyrosinemia type I, α1-antitrypsin deficiency, CF, and glycogen storage disease (type I). Although individually rare, together they account for approximately 10% of liver transplants in children.
Hemochromatosis/Iron Deposition.
Iron overload can occur by three basic mechanisms, including excess intestinal absorption, repeated blood transfusions, or inborn errors of metabolism. As systemic iron concentration overwhelms the capacity of ferritin to bind to and store iron, unbound iron accumulates in the cytoplasm of cells in the liver, spleen, pancreas, and bone marrow, resulting in cellular damage. A neonatal form of hereditary hemochromatosis known as neonatal iron storage disease exists, with the onset of liver disease in utero. Illness is usually evident within hours of birth, with features of fulminant hepatic failure.
Parenchymal alterations on both ultrasound and CT are considered nonspecific and nondiagnostic. MRI is the most useful noninvasive diagnostic tool for the identification of hepatic iron overload. As iron concentration in the hepatic parenchyma increases, signal intensity on T2 and gradient echo sequences decreases, leading to the appearance of a “black” liver [ Fig. 5.15 ]. Treatment of primary disease involves frequent phlebotomy, whereas treatment of secondary disease involves iron chelation therapy.
Tyrosinemia Type I.
Tyrosinemia type I is a rare disorder caused by a defect in the enzyme fumarylacetoacetate hydrolase. This defect leads to the accumulation of caustic metabolites in the liver and results in damage to DNA and a predisposition to the development of HCC. Patients may present with acute liver failure, chronic liver disease, or acute neurologic decompensation. In up to 42% of patients, clinical manifestations develop within the first year of life. Nephromegaly is frequently present. This entity should be considered when cirrhosis presents in early infancy. Treatment with 2-[2-nitro-4-(trifluoromethyl)benzoyl] cyclohexane-1,3-dione (NTBC) can be effective if started early. NTBC blocks the formation of caustic alkylating metabolites in the liver.
Glycogen Storage Disease.
Glycogen storage disease type I (von Gierke disease) is an autosomal recessive condition that results from a defect of the enzyme glucose-6-phosphatase, with resultant accumulation of glycogen in the liver, kidneys, and intestines. This disease may manifest in infancy with hypoglycemia, hepatomegaly, and nephromegaly. Hepatic adenomas and HCC have been described.
Ultrasound and CT most commonly show hepatomegaly and heterogeneous hepatic architecture. Nodules rarely occur and may appear hyperdense or hypodense on CT.
Hepatic Neoplasms
Primary hepatic tumors are relatively uncommon, accounting for 0.5% to 2% of all pediatric neoplasms. As in the adult population, the most common neoplasm involving the liver is metastatic disease, usually from neuroblastoma, Wilms tumor, or lymphoma. Malignant lesions account for approximately two-thirds of all primary liver tumors in children. A focused differential diagnosis for a liver tumor can be generated based on the age of the patient, serum tumor markers such as α-fetoprotein, and imaging characteristics.
Benign Lesions
Hemangioma.
Hemangioma is the most common hepatic mass presenting within the first 6 months of life, with nearly 50% of cases manifesting by the age of 1 month. Girls are affected more often than boys. This is a benign vascular tumor consisting of proliferation of endothelium-lined vascular spaces. Concomitant cutaneous hemangiomas occur in up to 50% of affected individuals. Hemangiomas are classified as infantile or congenital. The infantile form typically begins to grow after birth and slowly involutes after the second year of life. Congenital hemangiomas are present at birth and further subclassified as rapidly involuting congenital hemangiomas that regress within the first 14 months of life and noninvoluting congenital hemangiomas, which follow a clinical course similar to that seen with the infantile form.
Most hemangiomas present as a painless upper abdominal mass. However, serious clinical complications may occur. High-output congestive heart failure may be seen with high-flow lesions. This may be exacerbated by hypothyroidism caused by high levels of tumor-produced iodothyronine deiodinase. Occasionally, the Kasabach-Merritt syndrome of intratumoral consumptive coagulopathy may be seen. α-Fetoprotein levels are rarely elevated with this tumor.
The imaging appearance of hepatic hemangiomas may be quite variable. Multifocal lesions are typically small and relatively uniform in appearance. Large focal lesions are often heterogeneous, with areas of hemorrhage, infarction, and dystrophic calcification. Diffuse lesions result in marked enlargement of the liver, with replacement by large, vascular masses [ Fig. 5.16 ]. Hepatic arteries and veins are commonly enlarged, with prominent feeding and draining vessels within and surrounding the tumor(s). With bolus injection of contrast material and serial CT or MRI at the level of a lesion, early centripetal enhancement with delayed central enhancement over time will be observed.