Proto-oncogenes are components of signaling networks that act as positive growth regulators in response to mitogens, cytokines, and cell-to-cell contact. A gain-of-function mutation in only one copy of a proto-oncogene results in a dominantly acting oncogene that often fails to respond to extracellular signals.
Tumor suppressor genes are also components of the same signaling networks as protooncogenes, except that they act as negative growth regulators. They modulate proliferation and survival by antagonizing the biochemical functions of proto-oncogenes or responding to unchecked growth signals. In contrast to oncogenes, inactivation of both copies of tumor suppressor genes is required for loss of function in most cases.
DNA stability genes form a class of genes involved in both monitoring and maintaining the integrity of DNA. Loss of these genes results in defective sensing of DNA lesions as well as improper repair of the damaged template.
or gene changes in the three groups of genes described previously, may occur spontaneously because of random errors or result from exposure to agents as diverse as chemical mutagens, ionizing radiations, ultraviolet light, and viruses; and provide a growth or survival advantage that allows the cells to become the clonal origin of the tumor. To summarize, tumor evolution results from the accumulation of gene mutations that arise in a single cell that has suffered a disruption in its regulatory mechanisms for proliferation, self-elimination, immortalization, and genetic stability. This is illustrated in Figure 18.1.
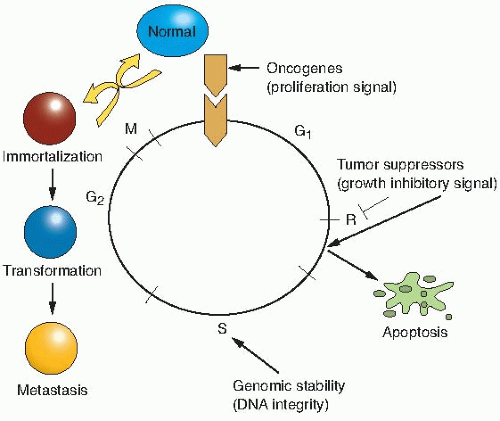 FIGURE 18.1 The process of malignant transformation results from mutations in three groups of genes: gain-of-function mutations that activate oncogenes, loss-of-function mutations that inactivate tumor suppressor genes, and loss of activity of DNA stability (e.g., repair) genes that increases the probability for genomic instability. This figure depicts how the stimulatory effects of oncogenes on the cell cycle are opposed by the inhibitory effects of tumor suppressor genes on the cell cycle that can lead to apoptosis. R indicates the restriction point that is regulated by the p53 and pRb tumor suppressor genes. The consequences of oncogene activation and tumor suppressor gene and DNA integrity gene inactivation are immortalization, transformation, and metastasis. |
with the advent of molecular biology, several groups identified the etiological agent for sarcoma formation in chickens as an RNA virus, designated as the Rous sarcoma virus (RSV), which belongs to a group of viruses designated as retroviruses—viruses whose genomes are composed of RNA. Thus, oncogenes were first discovered from a study of retroviruses that cause cancers in animals. Although the virus had been identified, it still remained to be elucidated how this retrovirus causes a sarcoma because another virus belonging to this same group of RNA viruses, avian leukosis virus (ALV), does not transform cells in culture or induce sarcomas. Analysis of the genomes of ALV and RSV revealed that RSV contains approximately 1,500 more base pairs (bp) of DNA than ALV. It was hypothesized, therefore, that these extra base pairs of DNA in the RSV genome are responsible for the tumorigenic activity. This was supported by an observation that deletion mutants of RSV that are missing this 1,500-bp region lose their transformation potential, but can still replicate and produce viral progeny normally. This led to the conclusion that the transforming activity and replicative activity of RSV are encoded by genetically distinct regions of the virus and that only a small portion of the RSV genome is needed for transformation.
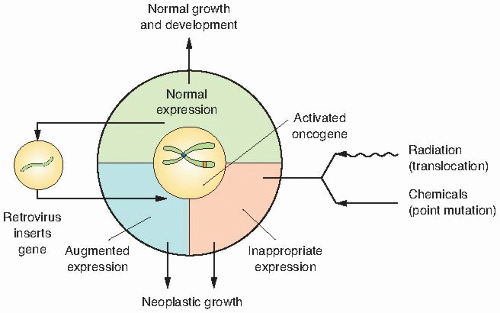 FIGURE 18.2 How the concept of oncogenes provides a ready answer for how agents as diverse as viruses, radiations, and chemicals all can induce tumors that are essentially indistinguishable one from another. The retrovirus inserts a gene; a chemical may activate an endogenous oncogene by a point mutation; radiation may do the same by causing, for example, a translocation. (Adapted from Bishop JM. Cellular oncogene retroviruses. Ann Rev Biochem. 1983;52:301-354, with permission.) |
Cancer can be caused by a genetically transmissible agent—in the case of chicken sarcoma, by a retrovirus containing a unique piece of genetic information that was later designated as the src gene;
Only a certain region of a retrovirus is needed for transformation; and
The region of the viral genome necessary for transformation is not involved in the normal replicative life cycle.
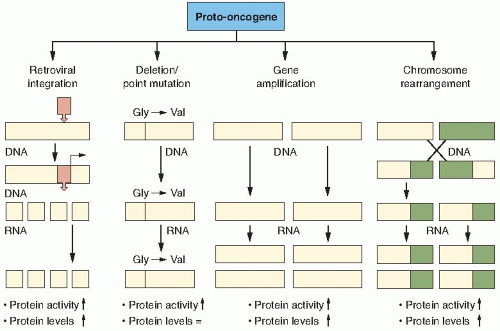 FIGURE 18.3 Four basic mechanisms of how a proto-oncogene can become an activated oncogene. Retroviral integration in proximity to a proto-oncogene results in transcriptional control of the proto-oncogene by the viral promoter, resulting in increased proto-oncogene protein and activity. Deletion or point mutations in the proto-oncogene result in increased activity of the proto-oncogene without necessarily changing transcription or protein levels of the proto-oncogene. Increased copy number of a proto-oncogene by gene amplification results in increased transcription, protein levels, and activity of the proto-oncogene. Chromosome translocation results in an altered proto-oncogene product that can have increased transcription, protein levels, and activity. All of these alterations in a proto-oncogene that occur at the DNA level manifest themselves at the protein level as increased activity and, in some cases, increased protein levels. |
deregulated expression by the viral LTR increases its expression and promotes cell growth.
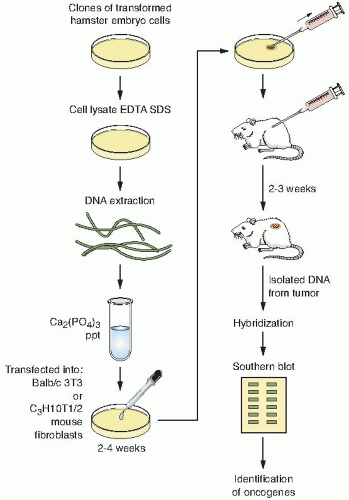 FIGURE 18.4 Schematic diagram of a typical DNA transfection protocol in which oncogenes can be isolated from cells transformed in vitro by either radiation or chemical carcinogens. DNA sequences are then characterized using Southern blot hybridization. |
The human DNA containing the transforming oncogene is transfected into mouse cells.
The DNA from the transformed mouse cells is serially transfected to reduce the amount of human DNA that is not associated with the transforming oncogene.
After several rounds of transfection, the DNA is isolated from a soft agar colony and digested with restriction enzymes to make a genomic DNA library.
The library is then screened with a human-specific repetitive probe that does not crossreact with the mouse’s DNA, thereby identifying human sequences in a mouse background.
Clones that possess human repetitive sequences are then isolated and digested with restriction enzymes to identify a similar-length fragment that is common to all transformants.
Finally, the DNA from the clones is transfected into mouse cells to confirm its oncogenic potential. If the oncogene is present in this genomic clone, then a significant percentage of the transfected mouse cells should be transformed when compared with transfecting genomic DNA from untransformed cells.
map near translocation breakpoints, as markers to identify potential translocation partners. Although numerous translocation breakpoints have been identified in hematopoietic neoplasms, few consistent translocations have been found in solid tissue tumors. The reason for this is still unclear, but may be attributed to the fact that hematopoietic cancers require fewer alterations for neoplasia than solid tumors. Table 18.1 provides examples of the chromosomal changes that result in oncogene activation and the associated human malignancies. Interestingly, there are no known examples of oncogenes activated by retroviruses in human malignancies.
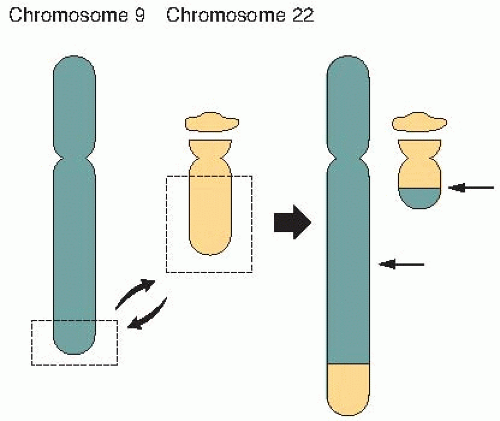 FIGURE 18.5 A symmetric translocation between chromosomes 9 and 22 brings together the bcl and abl genes to form a fusion gene associated with over 90% of cases of chronic myelogenous leukemia (CML). |
TABLE 18.1 Examples of Chromosomal Changes Leading to Oncogene Activation and Their Associated Murine or Human Malignanciesa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
patients with sporadic retinoblastoma. Based on these observations, Knudson proposed that in the inherited form of retinoblastoma, patients possess a germ line mutation of the retinoblastoma (Rb) gene in all the cells of their body, but inactivation of one Rb allele does not give rise to retinoblastoma. Thus, the disease appeared to be autosomal dominant because individuals are born with one mutated allele. However, a second mutation in a retinal cell is required to develop retinoblastoma. In the sporadic form of the disease, an individual has to acquire two Rb mutations in the same retinal cell to develop retinoblastoma. The “two-hit hypothesis” by Knudson provided a genetic basis to understand the differences in inherited and sporadic mutations in the onset of tumors and to advance the concept that both alleles of a tumor suppressor gene need to be inactivated to promote tumor development. Thus, tumor suppressor genes are recessive genes that require the inactivation of both functional gene copies before malignancies develop, whereas loss of one functional copy results only in increased cancer susceptibility. In addition, it is often the case that the same tumor suppressor gene involved in hereditary cancer syndromes, such as retinoblastoma, is also inactivated in other forms of cancer. The Rb gene itself has now been implicated in several other human cancers, which indicates that it may play a generalized role in tumor growth suppression in various tissues. For example, patients who are cured of familial retinoblastoma are at increased risk of osteosarcoma, small cell lung cancer, and breast cancer; although the loss of the Rb gene alone is sufficient for retinoblastoma, further changes are required for the development of these other tumors.
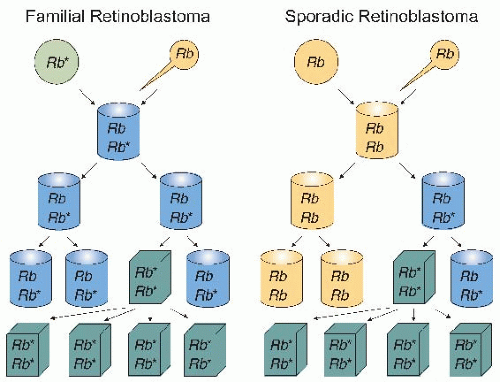 FIGURE 18.6 Rb mutations in familial and sporadic retinoblastoma. In familial retinoblastoma, one normal allele (Rb) and one mutated allele (Rb*) are inherited from either parent, resulting in a heterozygous individual containing Rb/Rb* retinal cells. Subsequent mutation in any retinal cell inactivates the remaining normal Rb allele, leading to loss of growth control and expansion of the homozygous mutant Rb*/Rb* retinal cells that leads to retinoblastoma. In sporadic retinoblastoma, two normal Rb alleles are inherited from each parent. First, a mutation inactivates one copy, resulting in heterozygous Rb/Rb* retinal cells. A subsequent mutation within the same retinal cell inactivates the remaining copy of normal Rb, leading to loss of growth control and expansion of homozygous Rb*/Rb* retinal cells that leads to retinoblastoma. (Illustrating the concepts proposed by Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820-823.) |
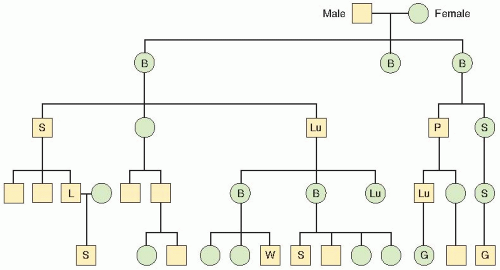 FIGURE 18.7 Pedigree analysis of familial cancer history of LFS. Symbols: B, breast cancer; G, glioblastoma; L, leukemia; Lu, lung cancer; P, pancreatic cancer; S, sarcoma; W, Wilms tumor. (Adapted from Li FP, Fraumeni JF. Prospective study of a family cancer syndrome. J Amer Med Assoc. 1982;247:2692-2694, with permission.) |
by the finding that mice lacking BRCA1 have the same phenotype as those null for the essential homologous recombination protein RAD51. Functionally, BRCA1 acts as a sensor of DNA damage and replication stress and mediates homologous recombination through BRCA2. In response to DNA damage-induced ionizing radiation, BRCA1 is phosphorylated at numerous sites by ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related (ATR), and checkpoint kinase 2 (Chk2) that direct it to associate with other repair proteins in nucleotide excision repair (NER), including xeroderma pigmentosum, complementation group C (XPC) and DNA damage-binding protein 2 (DDB2), in mismatch repair (MMR) MSH2, MSH6, and in DNA damage signalling ATM and the MRN complex (MRE11, Rad50, and NBS1). In contrast to BRCA1, BRCA2 appears to play a more direct role in homologous recombination by binding to RAD51 and aiding in the formation of RAD51 foci at the sites of DNA breaks. Cells from patients deficient in BRCA1 or BRCA2 are defective in homologous recombination and rely on error prone nonhomologous end- joining (NHEJ) to repair their DNA double strand breaks (DSBs). The resulting accumulation of mutations in BRCA1 and BRCA2 deficient cells promotes tumor formation.
TABLE 18.2 Examples of Cancer Predisposition Genes and Their Associated Syndromes | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
astrocytomas in which somatic homozygosity is observed for chromosome 10 in grade II and III astrocytoma and for both chromosomes 10 and 17 for grade IV glioblastoma.
 FIGURE 18.8 The process of somatic homozygosity. In a normal cell, there are two copies of each chromosome, one inherited from each parent. For a given suppressor gene to be inactivated, the copy must be lost from both chromosomes. This could, of course, occur by independent deletions from the two chromosomes; but in practice, it is more common for a single deletion to occur in one chromosome while the second chromosome is lost completely. The remaining chromosome, with the deletion, then replicates. The cell is thus homozygous, rather than heterozygous, for that chromosome. |
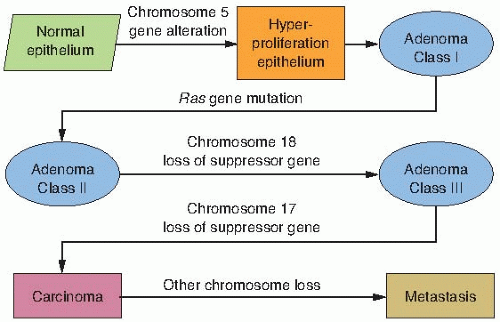 FIGURE 18.9 Cancer has long been thought to be a multistep process and has been described with operational terms such as initiation, promotion, and progression. In at least one human malignancy, namely, colon cancer, the molecular events during the progress of the disease have been identified. (Based on the work of Vogelstein and Kinzler.) |
 FIGURE 18.10 Illustrating the multistep nature of carcinogenesis and the concept of the mutator phenotype. The first step in carcinogenesis by radiation or any other agent may be a mutation in one of the gene families responsible for the stability of the genome. This may be a DNA repair gene, an MMR gene, or a gene in a family as yet unidentified. This leads to the mutator phenotype, with multiple mutations possible in both oncogenes and tumor suppressor genes. This then leads to a series of steps that result in an invasive metastatic cancer. Not all the same mutations need to be present in every case. |
residues so that they no longer require their respective growth factor (ligand) to signal induction of their kinase activity. Growth factor receptors can structurally be divided into extracellular ligand-binding domains (LBDs), transmembrane- spanning domains (TMS), and intracellular kinase domains. Although mutations have been found in all three domains, mutations in the ligand-binding domains are a common alteration that results in constitutive kinase activity that transduces the signal for the cell to proliferate. In addition to structural alterations in the receptor, some tumors overexpress growth factor receptors that make them hyperresponsive to physiologic levels of growth factor stimulation. In contrast to mitogenic-responsive growth factor receptors, a second class of receptors that transmit signals from the extracellular matrix can also regulate proliferation. Integrin receptors are the prototypical example of this class of regulators that transmit signals from different components of the extracellular matrix (ECM) to signal proliferation or quiescence.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


