FIGURE 16.1: Four-chamber view obtained by cross-sectional imaging of the chest. The four chambers are readily visualized: right atrium (RA), left atrium (LA), right ventricle (RV), left ventricle (LV). In addition, the two atrioventricular valves are seen: tricuspid valve (TV), and mitral valve (MV). The heart is perpendicular to the plan of imaging, and the ventricular septum is well visualized and appears intact. The patent foramen ovale (PFO) is also visualized.
Associated Anomalies: Most major types of CHD can be associated with an ASD, including endocardial cushion defect, anomalous pulmonary venous return, tetralogy of Fallot, double outlet right ventricle, and hypoplastic left heart syndrome. These anomalies will be discussed later in the chapter. Specific genetic defects are also associated with an atrial-level shunt. Holt–Oram syndrome consists of a secundum ASD, hypoplasia of the thumbs and radius, triphalangeal thumbs, abrachia, and phocomelia. Other congenital syndromes in which a secundum ASD may occur include Noonan syndrome, Treacher Collins syndrome, and the thrombocytopenia–absent radii (TAR) syndrome.
Treatment/Outcome: Postnatally, small secundum ASDs can be followed clinically as these defects often spontaneously close. Secundum ASDs that do require intervention are often amenable to percutaneous device closure in the cardiac catheterization laboratory, although some require surgical intervention, depending on size and location. All other types of ASDs require surgical closure, although timing is dependent on exact diagnosis, clinical situation, and associated cardiac abnormalities.
Ventricular Septal Defects
Definition: A ventricular septal defect (VSD) is a defect in the ventricular septum that causes a communication between the two ventricles.
Incidence: VSDs are the most commonly diagnosed CHD, involving 25% of live births with CHD.1
Pathology and Hemodynamics: Like ASDs, VSDs are classified by their location within the ventricular septum, though various classification systems exist. VSDs can be classified as muscular, atrioventricular, conoventricular (including membranous/perimembranous and malalignment-type defects), and conoseptal. Muscular VSDs are defects within the muscular septum and are entirely surrounded by muscle. These defects can be further classified by location within the muscular septum, such as apical (below the moderator band), midmuscular, posterior, and anterior. Atrioventricular (AV) (or inlet) defects are defects in the posterior ventricular septum abutting the AV valves. These defects may or may not be associated with an atrioventricular canal defect. Membranous defects involve the membranous septum with the defect bordering the tricuspid valve. Perimembranous defects are around the membranous septum. Conoventricular VSDs encompass those defects that involve the conal or outlet septum. The malalignment-type VSD is a type of conoventricular VSD. Defects of conal septal position result in malalignment, yielding the potential for outflow tract obstruction. The conal septum can be anteriorly displaced (as in tetralogy of Fallot) or posteriorly displaced (often associated with interrupted aortic arch). Defects of the conal septum resulting in conal hypoplasia are closely associated with the semilunar valves. These defects can lead to aortic insufficiency due to lack of support of the aortic valve and distortion of the right coronary cusp if the valve gets pulled into the defect.
Associated Anomalies: Most VSDs are isolated, but they can be found with almost all other CHD. Therefore, when a VSD is identified, all other cardiac structures must be thoroughly evaluated (i.e., to exclude more extensive anomalies, such as tetralogy of Fallot and coarctation of the aorta). VSDs are also seen in fetuses with chromosomal abnormalities, including trisomy 21, 18, and 13.
Imaging: VSDs are best seen when the ultrasound beam is perpendicular to the septum, as “false dropout” occurs when the beam is parallel to the septum (Fig. 16.2). Large defects will be easily visible; however, small defects are often missed. Color Doppler demonstrates bidirectional shunting. Perimembranous defects will be wedged under the tricuspid valve; these types of defects are best seen from the short- and long-axis views of the heart. Conal septal defects occur in the outlet septum in the muscle that supports the aortic valve adjacent to the pulmonary valve. These defects are best seen in short axis imaging. Anterior malalignment of the conal septum is typically seen in conjunction with aortic override, making the diagnosis of tetralogy of Fallot. The conal septum may also be posteriorly malaligned, which may be associated with subaortic narrowing, aortic valve stenosis, and coarctation of the aorta or interrupted aortic arch. Atrioventricular canal–type VSDs or inlet defects are well seen in the four-chamber view under the atrioventricular valves.
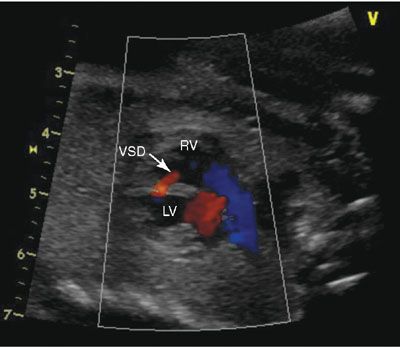
FIGURE 16.2: A sagittal image of the heart, sweeping leftward; one can see the anterior right ventricle (RV) and the posterior left ventricle (LV). The red flow represents a muscular ventricular septal defect (VSD). The blue color flow demonstrates the laminar flow in the right ventricular outflow tract.
Treatment/Outcome: Postnatally, small VSDs can be followed clinically as perimembranous and muscular defects often close spontaneously (more likely when the defects are smaller than 3 mm in a term infant). Moderate-to-large defects often require surgical closure in the setting of congestive heart failure that cannot be managed medically. In older patients, muscular VSDs are sometimes amenable to percutaneous device closure in the cardiac catheterization laboratory, although most VSDs that require closure necessitate surgical intervention. Malalignment and atrioventricular canal–type defects will not close spontaneously and therefore need surgical closure. Typically, conal septal hypoplasia defects do not close, but the decision to operate depends on the amount of left to right shunting and the effect the defect has on the aortic valve.
Endocardial Cushion Defects
Definition: Endocardial cushion defects include the spectrum of diseases involving the endocardial cushion and include complete atrioventricular canal (CAVC), transitional AVC defects, and isolated primum ASD with cleft mitral valve. These defects can be classified by size/presence of the VSD, the balance of the atrioventricular valve over the ventricles, and the anatomy of the common valve.
Incidence: Endocardial cushion defects account for 5% to 7% of all live births with CHD.1
Pathology and Hemodynamics: A CAVC defect includes defects of the atrial septum (primum ASD) and ventricular septum (atrioventricular VSD) with a common atrioventricular valve. A partial AVC is comprised of a primum ASD and a cleft mitral valve. A transitional AVC is similar to a complete CAVC except that there is a small restrictive VSD. CAVCs can be further classified by the anatomy of the anterior bridging leaflet of the common atrioventricular valve. Using the classification system of Rastelli et al.,2 in type A defects, the anterior bridging leaflet is divided and has attachments to the crest of the ventricular septum. In type B defects, the anterior bridging leaflet is partially divided, but with no attachments to the ventricular septum. In type C defects, the anterior bridging leaflet is both undivided and unattached to the ventricular septal crest. The Rastelli classification is best determined by short axis imaging of the ventricles, noting the valve en face.
Further classification of endocardial cushion defects must include a description of the absolute and relative size of the ventricles. In a balanced CAVC, both ventricles are approximately equal in size, and the common valve is positioned symmetrically over both ventricles. In an unbalanced CAVC, the atrioventricular canal sits predominantly over either the right or the left ventricle; flow is toward that ventricle, and the contralateral ventricle is usually small.
Associated Anomalies: Defects associated with endocardial cushion defects include tetralogy of Fallot, double outlet right ventricle, coarctation of the aorta, subaortic stenosis, pulmonary valve stenosis, and single ventricle. In the cases of multiple defects (i.e., CAVC with double outlet right ventricle and/or with complete heart block [CHB]), heterotaxy syndrome should be considered. When CAVC exists as an isolated lesion, trisomy 21 is present in 60% of the cases.3 Endocardial cushion defects may also occur with T13, T18, and T21.
Imaging: Endocardial cushion defects are easily seen in the four-chamber view, with the components of the ASD, VSD, and common valve easily visible (Figs. 16.3 to 16.5). The details of the common atrioventricular valve (en face view) are best seen in short axis imaging of the ventricle (Fig. 16.6). The cleft of the anterior leaflet refers to the division of the leaflet with the attachment to the left ventricular septal surface. Color Doppler should be performed to evaluate for atrioventricular valve regurgitation in the four-chamber view.
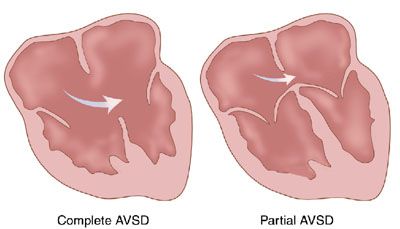
FIGURE 16.3: Illustration of partial and complete atrioventricular canal. (From Rice MJ, McDonald RW, Pilu G, et al. Cardiac malformations. In: Nyberg DA, McGahan JP, Pretorius DH, et al, eds. Diagnostic Imaging of Fetal Anomalies. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:459.)
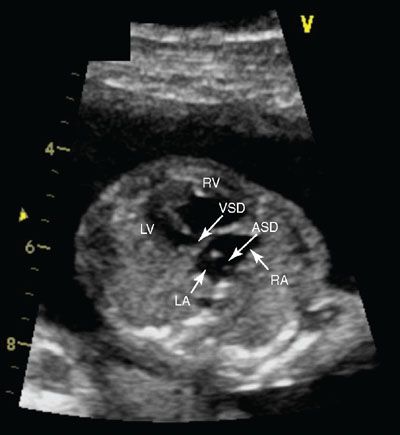
FIGURE 16.4: Four-chamber view of a complete atrioventricular canal. The crux of the heart is absent in this view, demonstrating both the inlet ventricular septal defect (VSD) (arrow) and the primum atrial septal defect (ASD) (arrow). The patent foramen ovale is also present in this image. In this systolic view, the single or common valve is also appreciated. RA, right atrium (arrow); LA, left atrium; RV, right ventricle (arrow); LV, left ventricle.
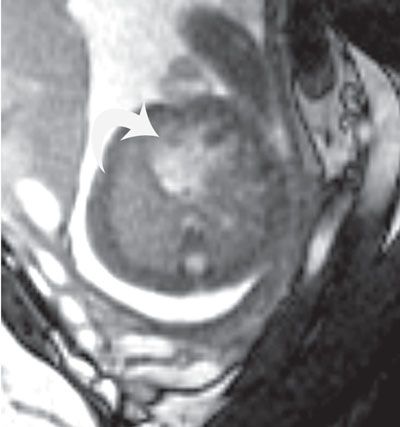
FIGURE 16.5: Atrioventricular canal. Axial SSFP MR image of the fetal heart demonstrates a single common valve (curved arrow). A large primum atrial septal defect and ventricular septal defect are also present.
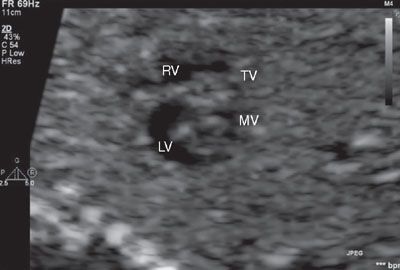
FIGURE 16.6: In this short axis view of the fetal heart, the two components of the right and left side of the atrioventricular valves are well seen. This image will aid in determining Rastelli classification and balance of the valves over the ventricles. Here the right-sided atrioventricular valve is labeled as the tricuspid valve (TV), and the left-sided atrioventricular valve is labeled as the mitral valve (MV). RV, right ventricle; LV, left ventricle.
Treatment/Outcome: CAVC defects always require surgical correction, although the type of repair can be dependent on other anatomic factors (e.g., a significantly unbalanced CAVC may require single ventricle palliation). The technique of the surgical repair of CAVCs can vary widely, and be comprised of a single patch, two-patch, or modified patch technique. Short- and long-term outcomes are greatly dependent on associated cardiac defects, residual atrioventricular valve regurgitation, outflow tract obstruction, and chromosomal anomalies.
Inflow Defects
Diseases of the Tricuspid Valve
Ebstein Anomaly of the Tricuspid Valve
Definition: Ebstein anomaly of the tricuspid valve is defined by specific anatomical features, including apical displacement of the septal and posterior leaflets of the tricuspid valve within the right ventricle (Fig. 16.7). This creates a displaced tricuspid valve orifice into the body of the ventricle with the proximal right ventricle becoming functionally part of the right atrium.
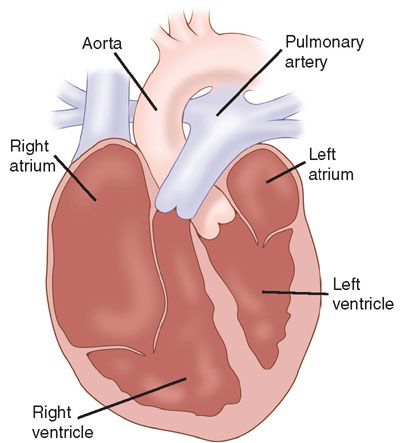
FIGURE 16.7: Illustration of Ebstein anomaly of the tricuspid valve. The tricuspid valve is displaced into the right ventricle. (From Rice MJ, McDonald RW, Pilu G, et al. Cardiac malformations. In: Nyberg DA, McGahan JP, Pretorius DH, et al, eds. Diagnostic Imaging of Fetal Anomalies. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:461.)
Incidence: Ebstein anomaly accounts for 0.5% of live births with CHD, although it occurs in approximately 3% to 8% of fetal CHD, with a 44% rate of fetal demise.1
Pathology and Hemodynamics: There is a wide spectrum of disease related to Ebstein anomaly, ranging from minimal displacement of the tricuspid valve to severe apical displacement wherein nearly the entire right ventricle is “atrialized” and there is little to no functional right ventricle. Severe tricuspid regurgitation can be associated with secondary hydrops fetalis and in utero fetal demise. In some cases, there is associated pulmonary stenosis or atresia.
Associated Anomalies: Ebstein anomaly may be associated with other CHD, such as severe pulmonary stenosis or pulmonary atresia, ASDs, Wolf–Parkinson–White syndrome and congenitally corrected transposition of the great arteries.4 Additionally, severe cardiomegaly may contribute to lung hypoplasia, increasing the risk of death.
Imaging: Ebstein anomaly is well seen in the four-chamber view (Fig. 16.8). The displacement of the hinge point of the septal leaflet of the tricuspid valve is quantified by measuring the distance between the crux of the heart (from the mitral valve hinge point) and the hinge point of the septal leaflet of the tricuspid valve; this distance should be less than 5 mm at birth. Cardiomegaly secondary to atrial or ventricular dilation may be present. Color Doppler should be used to evaluate the degree of tricuspid regurgitation (Fig. 16.9). Tricuspid regurgitation may progress during fetal life. When severe tricuspid regurgitation exists, the right ventricle may not be able to generate sufficient pressure to open the pulmonary valve even though it may be patent; this is termed functional pulmonary atresia. The branch pulmonary arteries can be best seen in short axis imaging; these may be normal in size or hypoplastic. The ductus arteriosus flow may be normal or reversed. If flow is reversed, associated “functional” or anatomical pulmonary atresia is likely present. Finally, the right heart may be dilated, causing the left heart to be compressed and appear hypoplastic.
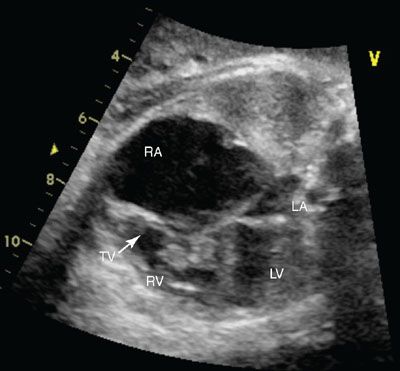
FIGURE 16.8: Four-chamber view of Ebstein anomaly of the tricuspid valve in systole. Note the severely enlarged right atrium (RA) and the abnormal tricuspid valve (TV). LA, left atrium; RV, right ventricle; LV, left ventricle.
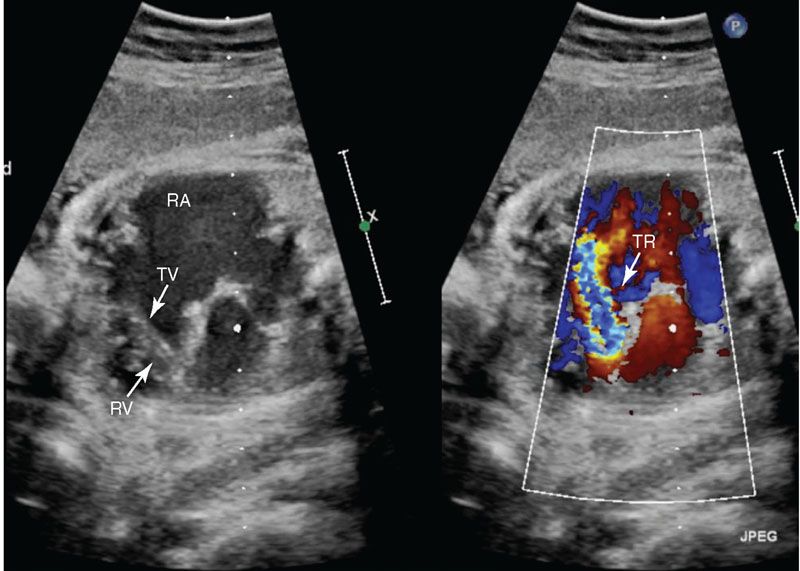
FIGURE 16.9: Four-chamber color compare view of Ebstein anomaly of the tricuspid valve in diastole. The apical displacement of the tricuspid valve leaflets (TV) into the right ventricle (RV) is readily visualized. There is moderate to severe tricuspid insufficiency (TR) with severe right atrial (RA) enlargement.
Prognosis: A neonate with significant Ebstein anomaly associated with pulmonary stenosis or atresia will present with cyanosis in the immediate newborn period. Postnatally, when the pulmonary vascular resistance falls, antegrade pulmonary blood flow may improve if the valve is not atretic. If there is pulmonary atresia or a delayed decrease in pulmonary vascular resistance, prostaglandin is often used to maintain ductal patency and pulmonary blood flow. These neonates may need a surgical systemic to pulmonary artery shunt to ensure adequate pulmonary blood flow. Additional surgeries such as right ventricular outflow patch reconstruction or single ventricle palliation may need to be performed, depending on the degree of right ventricular outflow tract obstruction, right ventricular hypoplasia, or tricuspid valve competency. Alternatively, if the tricuspid valve displacement is minimal and the tricuspid valve regurgitation is mild, patients may not be diagnosed until adulthood (often due to associated atrial arrhythmias).
Tricuspid Valve Dysplasia: Tricuspid valve dysplasia is a nonspecific term that can include various abnormalities of tricuspid valve anatomy. Unlike Ebstein anomaly, in tricuspid valve dysplasia the tricuspid valve leaflet attachments are at the level of the valve annulus. The leaflets are often thickened with poor coaptation and have abnormal papillary muscle and chordae. Various amounts of tricuspid valve regurgitation will often be present, and the degree of the regurgitation will determine severity and outcome of the disease. Tricuspid valve dysplasia is associated with varying degrees of right ventricular hypoplasia, pulmonary stenosis, or pulmonary atresia.
Isolated Tricuspid Valve Regurgitation: Tricuspid valve regurgitation can vary in severity and clinical significance. An attempt to quantify the regurgitation can be made by evaluating the duration of the insufficiency during systole, the peak velocity of the regurgitant jet, the diameter of the regurgitant jet, and how far back within the right atrium the jet reaches.5 Trivial tricuspid valve regurgitation may be found in up to 5% of fetuses with structurally normal hearts, and resolution is usually expected.6 More significant amounts of tricuspid valve regurgitation can be associated with structural heart defects, volume overload states (such as fetal anemia or vein of Galen aneurysms), poor fetal cardiac function, or in fetuses of maternal anti SSA/SSB antibodies (SSA) antibody positive women.7
Cor Triatriatum Dexter: Cor triatriatum dexter is an extremely rare condition whereby there is a persistence of a right venous valve that results in septation of the smooth and trabeculated portions of the right atrium. Normal venous valves exist to help direct oxygenated blood flow from the placenta across the foramen ovale. Complete persistence of the right venous valve or prominent chiari network may result in cor triatriatum dexter. The physiology and presentation is dependent on the degree of septation of the right atrium. Cor triatriatum dexter can occur in isolation or with other right-sided defects, such as pulmonary stenosis or atresia, tricuspid valve anomalies, ASD, or Ebstein anomaly.
Tricuspid Stenosis and Tricuspid Atresia: Tricuspid stenosis and tricuspid atresia are typically associated with varying degrees of right ventricular hypoplasia. Either of these lesions may be associated with a VSD. It is important to evaluate the size of the right ventricle and the source of pulmonary blood flow (antegrade via a pulmonary artery or retrograde via a ductus arteriosus). These lesions are typically classified in the single ventricle lesion and will be discussed further in the single ventricle section.
Diseases of the Mitral Valve
Dysplastic Mitral Valve: A dysplastic mitral valve with primary mitral regurgitation is rare. Mitral regurgitation is more often associated with severe aortic stenosis and left ventricular injury, endocardial cushion defects, or primary left ventricular heart failure. Mitral regurgitation is also seen in fetuses of SSA antibody positive mothers or in those with myocarditis. Often, the left ventricle is dilated, and the endocardium and papillary muscles are echo bright.
Mitral Stenosis: Congenital mitral stenosis is a rare condition. Congenital mitral stenosis can occur with other left-sided obstructive lesions. Shone syndrome is a term used for patients with multiple left-sided obstructions. Severe mitral stenosis may lead to progressive aortic and left ventricular hypoplasia in utero. In congenital mitral stenosis, the annulus, leaflets, chordae, and/or the papillary muscles may be abnormal.8 The chordae may be absent or extremely short so that the valve apparatus attaches directly to the papillary muscles; this is termed a mitral valve arcade. The chordae may attach to a single papillary muscle; this is termed a parachute mitral valve. Imaging of the heart in the short axis will demonstrate the papillary muscles. Imaging from the apical four-chamber view will demonstrate the mitral valve annulus diameter and chords. Color and pulse wave Doppler should be used to identify the degree of stenosis or regurgitation. Ventricular size and function should be assessed, as should the direction of foramen ovale flow. Reversed foramen ovale flow suggests severe disease. Some patients may be candidates for mitral valvuloplasty postnatally; however, patients are generally managed conservatively, and surgical intervention is delayed as long as possible valve replacement may be needed.
Cor Triatriatum Sinister: Cor triatriatum sinister describes a membrane within the body of the left atrium, proximal to the left atrial appendage, which causes a varying degree of obstruction to pulmonary venous return to the left side of the heart. Developmentally, this is thought to be due to incomplete incorporation of the pulmonary veins into the left atrium. There is usually a communication between the superior and the inferior chambers of the left atrium of variable size. There may be an ASD or PFO with the right atrium connecting to the distal chamber, and there may be anomalous pulmonary venous drainage. The physiology of this lesion depends on the degree of restriction between the superior and the inferior chambers caused by the cor triatriatum membrane. If the opening is large, the neonate will be asymptomatic. If the opening is small, there will be venous obstruction with subsequent pulmonary edema, pulmonary hypertension, and decreased cardiac output. The echocardiographic evaluation should include the size of the foramen ovale, the communication between the superior and the inferior chambers, and pulmonary venous return. Color and pulse wave Doppler should be performed to evaluate restriction of the communication. The treatment includes resection of the left atrial membrane if it is restrictive or in patients who are symptomatic or have pulmonary artery hypertension.
Supravalvar Mitral Ring: A supravalvar mitral ring consists of a ring of tissue that adheres to the atrial surface of the mitral valve leaflets. This tissue is distal to the left atrial appendage (thus differentiating it from a cor triatriatum membrane). This may be associated with other left-sided defects such as subaortic stenosis, coarctation of the aorta, and VSD. There will be varying degrees of mitral regurgitation and mitral stenosis. The membrane will be best seen in the long axis or posteriorly tipped in four-chamber views adherent to the mitral valve annulus. Surgical resection is the treatment of choice.
Outflow Defects
Pulmonary Valve Stenosis
Definition: Pulmonary valve stenosis is an abnormality in the pulmonary valve leading to varying degrees of obstruction to the right ventricular outflow tract, often due to fusion of the valve commissures.
Incidence: Isolated pulmonary valve stenosis accounts for approximately 7% of live births.1
Pathology and Hemodynamics: Pulmonary valve stenosis, like stenosis at any valve, can progress during gestation. Interval imaging should be performed as the disease can evolve. Serial fetal echocardiograms will allow for postnatal planning to assess for disease progression. Severe pulmonary stenosis may cause inadequate pulmonary blood flow and require postnatal initiation of prostaglandin to maintain patency of the ductus arteriosus.
Associated Anomalies: Complete fetal echocardiogram should include evaluation for commonly associated cardiac anomalies, such as an ASD, tetralogy of Fallot, Ebstein anomaly, and double outlet right ventricle. Associated genetic anomalies and syndromes should be considered, such as Noonan syndrome and Beckwith–Wiedemann. Rubella is a known teratogen causing both pulmonary stenosis and peripheral pulmonic stenosis.
Imaging: The pulmonary valve can be visualized from several different views and should be assessed for thickening, doming, and poor excursion of the valve leaflets. The amount and velocity of antegrade blood flow can be assessed with color and pulsed-wave Doppler. Pulse wave Doppler can be used to calculate the gradient across the valve using the modified Bernoulli equation, where the pressure gradient across a discrete stenosis is equal to 4 × (velocity2), also written as 4v2.3 Right ventricular hypertrophy and/or hypoplasia and tricuspid valve regurgitation may be seen in the four-chamber view. Right ventricular hypertension may develop and right ventricular pressures can be estimated by measuring the velocity of the tricuspid regurgitation and calculating right ventricular pressure by the Bernoulli equation where right ventricular pressure equals 4 × (tricuspid velocity)2.3 Poststenotic dilatation of the main pulmonary artery may be associated with pulmonary stenosis. In some cases, mild pulmonary stenosis may not be visualized, and the only indicator of valve disease is poststenotic dilation of the main pulmonary artery. In severe or critical pulmonary stenosis, there will be reversed flow in the ductus arteriosus.
Treatment/Outcome: The prognosis for pulmonary valve stenosis is dependent on the severity of the outflow tract obstruction and associated defects. Mild or moderate pulmonary stenosis has an excellent prognosis and often requires no immediate postnatal intervention, although percutaneous balloon valvuloplasty may be required in infancy or early childhood. More severe forms of pulmonary stenosis or functional pulmonary atresia may require more urgent neonatal intervention. Reversal of flow in the ductus arteriosus prenatally can be indicative of ductal-dependent pulmonary blood flow and need for early initiation of prostaglandin to maintain ductal patency until balloon valvuloplasty or surgical intervention in the immediate neonatal period can be undertaken.
Pulmonary Atresia
Definition: In pulmonary atresia, no antegrade flow is present across the pulmonary valve leaflets, and the leaflets are often fused (Fig. 16.10). The diagnosis of pulmonary atresia with intact ventricular septum is used to distinguish this disease from tetralogy of Fallot with pulmonary atresia.
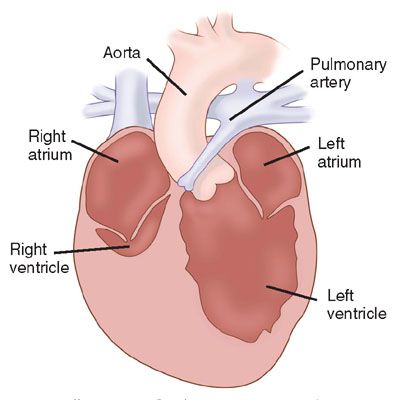
FIGURE 16.10: Illustration of pulmonary atresia with intact ventricular septum. (From Rice MJ, McDonald RW, Pilu G, et al. Cardiac malformations. In: Nyberg DA, McGahan JP, Pretorius DH, et al, eds. Diagnostic Imaging of Fetal Anomalies. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:464.)
Incidence: Pulmonary atresia with intact ventricular septum occurs in 1% to 2% of all live births with CHD.
Pathophysiology and Hemodynamics: The pulmonary valve is atretic in pulmonary atresia/intact ventricular septum. There are usually varying degrees of tricuspid valve and right ventricular hypoplasia. The pulmonary valve leaflets are often present but fused, and the branch pulmonary arteries are typically small but not severely hypoplastic. Coronary artery fistulous connections to the right ventricle may be present and are related to right ventricular pressure elevation. Some will have associated coronary artery atresia, obstruction, or right ventricular pressure-dependent circulation.
Imaging: In the four-chamber view, the tricuspid valve will be patent but is usually small. The right ventricle may be hypoplastic and echogenic (Fig. 16.11). There may be tricuspid insufficiency as there is no other egress from the right ventricle (Fig. 16.12). Use of pulse wave Doppler of the tricuspid insufficiency will allow an estimation of right ventricular pressure, which may be suprasystemic (Fig. 16.13). Imaging from the short axis and ductal views will demonstrate no antegrade flow across the pulmonary valve and reversed flow in the ductus arteriosus. A main pulmonary artery will be present and give rise to continuous though small pulmonary arteries. Finally, coronary fistulae may be present and are best visualized in either the four-chamber view or short axis of the ventricles with color Doppler (Fig. 16.14). Typically, there is a stretched PFO or secundum ASD.
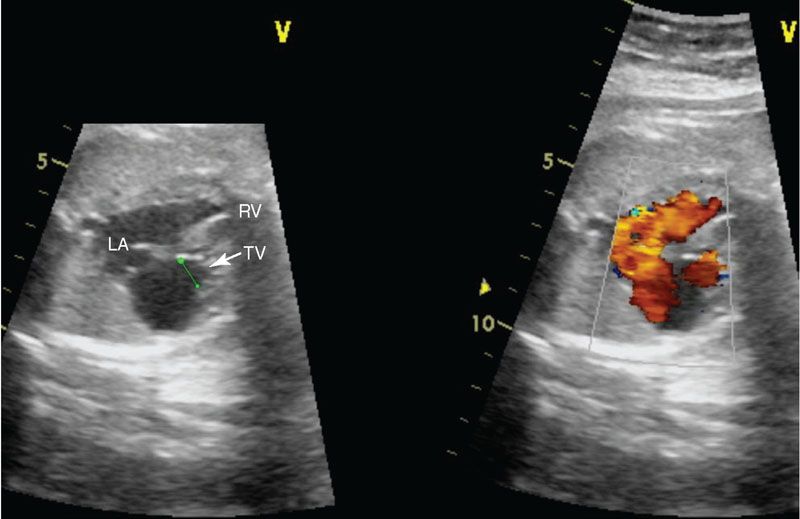
FIGURE 16.11: Color compare view of pulmonary atresia with intact ventricular septum demonstrating a hypoplastic tricuspid valve (TV) annulus. Note in the color image the foramen ovale is widely patent, allowing unrestrictive right to left atrial flow. LA, left atrium; RV, right ventricle.
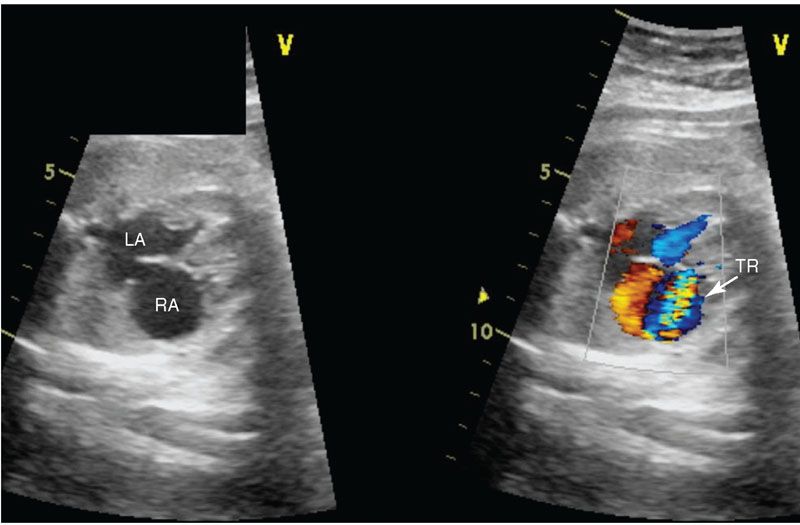
FIGURE 16.12: Color compare view of four-chamber view of fetal heart with pulmonary atresia intact ventricular septum. The blue color Doppler demonstrates severe tricuspid regurgitation (TR). LA, left atrium; RA, right atrium.
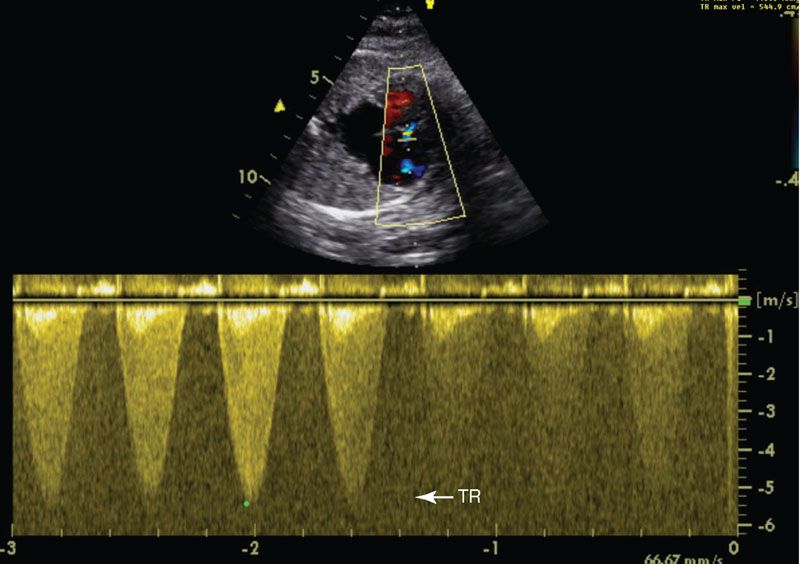
FIGURE 16.13: Pulse wave Doppler imaging of the tricuspid regurgitation (TR) demonstrates a high velocity, 5.3 m per second, predicting a right ventricular pressure of 112 mm Hg.
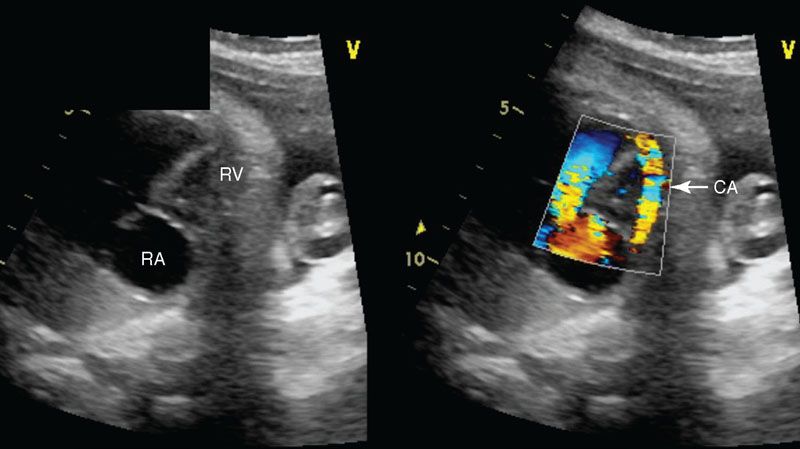
FIGURE 16.14: Color compare four-chamber view of pulmonary atresia intact ventricular septum with a fistula from the right ventricle (RV) to the coronary artery (CA). Color Doppler is necessary to show this diagnosis. RA, right atrium.
Treatment/Outcome: By definition, newborns with pulmonary atresia are ductal dependent, and therefore prostaglandin should be initiated immediately after delivery. In addition to echocardiography, cardiac catheterization is performed to evaluate coronary artery anatomy. Right ventricular–dependent coronary arteries are a significant risk factor for death. A small percentage of patients will have an adequate size tricuspid valve (>−2 to −3 z-score), a well-developed right ventricle, and normal coronary arteries with flow not dependent on the right ventricle. These patients may be candidates for radiofrequency ablation to open up the right ventricular outflow tract. The remainder of patients will need a systemic to pulmonary artery shunt. Patients are followed over the first year of life, and if the tricuspid valve z-score is >−2 and the right ventricle is adequate, a two ventricular repair may be undertaken later. If the right ventricle is small but has some output, a “one and a half ventricular repair” may be performed. This repair includes a Glenn shunt (superior vena cava anastomosis to the pulmonary artery) with the inferior vena cava flow returning to the right ventricle to be pumped to the lungs. Finally, some patients must follow a single ventricle approach to repair. One indication for single ventricle palliation is right ventricular–dependent coronary circulation. In these patients, the right ventricle cannot be used as the pulmonary pump because of the high risk of coronary artery ischemia. Other indications for single ventricle palliation is an extremely small tricuspid valve (z-score < −4) or severely hypoplastic right ventricle. In these patients, a systemic to pulmonary artery shunt will be placed in the newborn period. These patients will then ultimately undergo Glenn shunt and Fontan procedure (see hypoplastic left heart syndrome for further discussion of these surgeries).
A small subset of fetuses with pulmonary atresia/intact ventricular septum may be considered for fetal intervention; however, there is debate as to whether intervention should be offered.9–11 The goal of intervention is to promote right ventricular growth and avoid single ventricle palliation or as a lifesaving measure if severe tricuspid regurgitation and hydrops exists and there is impending fetal demise.12 The technique for intervention is difficult given the right ventricular cavity is small, hypertrophied, and located behind the sternum.13
Long-term prognosis for children with pulmonary atresia intact ventricular septum depends on the degree of tricuspid, degree of right ventricular hypoplasia, and the presence of coronary abnormalities. The outcome is good for those that undergo pulmonary valve dilation or right ventricular outflow plasty with a two ventricular repair. In patients that undergo single ventricle palliation, early deaths have been reported secondary to coronary ischemia, but long-term survival is reported to be 83% at 5 years.14
Aortic Stenosis
Definition: Aortic stenosis is an abnormality in the aortic valve leading to left-sided outflow tract obstruction at the level of the aortic valve. The valve may be bicuspid, unicuspid, or thickened with fused commissures.
Incidence: Aortic stenosis accounts for 5% to 7% of children with cardiac disease. A bicuspid nonstenotic aortic valve may be as common as 3% of the population but does not always present as heart disease in childhood.
Pathology and Hemodynamics: The abnormal aortic valve itself may have three leaflets; more commonly, there may be fusion of the leaflets, resulting in unicuspid or bicuspid valve. Aortic stenosis, like stenosis at any valve, can progress during gestation. Interval imaging should be performed as the disease can evolve. Serial fetal echocardiograms will allow for postnatal planning to assess for disease progression. Severe aortic stenosis may cause inadequate systemic blood flow and require postnatal initiation of prostaglandin to maintain patency of the ductus arteriosus.
Associated Anomalies: Aortic stenosis is often familial. Bicuspid aortic valve and coarctation of the aorta can be seen in association with Turner syndrome. Supravalvar aortic stenosis can be associated with Williams syndrome. Subvalvar aortic stenosis does not typically present in the fetus. Associated abnormalities include hypoplasia of the left ventricle, mitral valve, or aortic arch (see hypoplastic left heart syndrome section).
Imaging: Ultrasound findings vary according to the severity of the disease. The aortic leaflets will be best visualized in a short axis view, and they may appear thickened and fused. Color and pulse Doppler should be performed to accurately determine degree of stenosis (Fig. 16.15). Aortic flow gradient can be calculated by the Bernoulli equation (aortic gradient = 4 × velocity2). In the presence of severe left ventricular dysfunction, the gradient may be minimal. Dilation of the ascending aorta is a subtle clue of mild aortic stenosis and is best seen in the long axis evaluation of the left ventricular outflow tract. Significant mitral regurgitation may be present and is best visualized in the four-chamber view. Severe aortic stenosis may cause left ventricular dysfunction resulting in retrograde aortic arch and foramen flow; this may be a precursor to hypoplastic left heart syndrome.
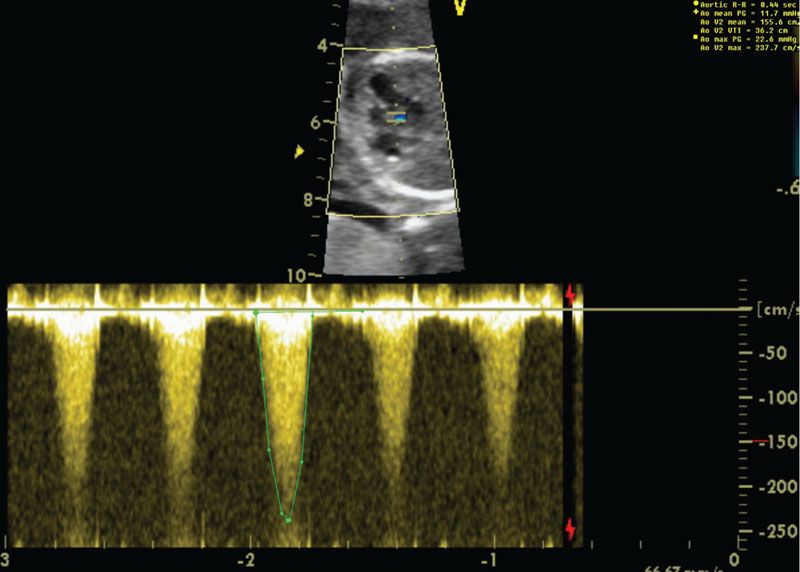
FIGURE 16.15: In the upper panel, the heart is imaged in a four-chamber view angled cranially. The aortic valve and left ventricle appear echobright. Pulse wave Doppler demonstrates increased velocity across the aortic valve of 4.1 m per second or a peak gradient of 67 mm Hg.
Treatment/Outcome: Postnatal management of aortic stenosis is dependent on severity of the disease. With critical aortic stenosis, there is little to no antegrade blood flow, and prostaglandin is necessary to supply sufficient cardiac output. Balloon aortic valvuloplasty is performed if there is aortic stenosis with ventricular dysfunction, or if the peak gradient is greater than 70 to 80 mm Hg. Balloon angioplasty is typically successful, although most patients will need further procedures and ultimate aortic valve replacement. Morbidity and mortality will be dependent on the degree of stenosis or left ventricular dysfunction.
Conotruncal Defects
Conotruncal defects encompass a wide spectrum of CHD with abnormalities of the connection between the ventricles and the great vessels, including tetralogy of Fallot, transposition of the great arteries, truncus arteriosus, and double outlet right ventricle. Conotruncal anomalies account for 20% to 30% of all live births with structural CHD.
Tetralogy of Fallot
Definition: The anatomic features of tetralogy of Fallot include infundibular and valvar pulmonary stenosis, a malalignment-type VSD, overriding aorta, and right ventricular hypertrophy (a late sequela of the outflow tract obstruction and not observed in utero) (Fig. 16.16). The aortic valve is in continuity with the right ventricle.
FIGURE 16.16: Illustration of tetralogy of Fallot. A large aorta overrides a ventricular septal defect, and the pulmonary outflow tract is hypoplastic. (From Rice MJ, McDonald RW, Pilu G, et al. Cardiac malformations. In: Nyberg DA, McGahan JP, Pretorius DH, et al, eds. Diagnostic Imaging of Fetal Anomalies. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:475.)
Incidence: Tetralogy of Fallot is the most common type of cyanotic CHD, accounting for approximately 9% of CHD live births.1
Pathophysiology and Hemodynamics: If not diagnosed prenatally, timing of presentation can vary significantly, depending on the severity of the right ventricular outflow tract obstruction. Postnatally, patients with tetralogy of Fallot become cyanotic because of right to left shunting across the VSD in the presence of severe right ventricular outflow tract obstruction. The degree of shunting and cyanosis is secondary to the right ventricular outflow tract obstruction. Newborns with little right to left shunting will be “pink” and will have clinical symptoms of a large VSD. The right ventricular outflow tract obstruction may progress and therefore warrants close observation. During a “tet spell,” there is dynamic muscular obstruction of the right ventricular outflow tract, causing severe cyanosis. “Tet spells” may be precipitated by crying, illness, or decreased systemic vascular resistance. Treatment includes oxygen, IV fluids, morphine, knee to chest position, and often emergency surgery.
Associated Anomalies: Tetralogy of Fallot is often associated with genetic and extracardiac anomalies such as DiGeorge syndrome, VACTERL (verterbral anomalies, anal atresia, cardiac anomalies, tracheoesophageal fistula, renal anomalies, and limb anomalies) association, and Goldenhar syndrome, among many others. A right aortic arch suggests a deletion on chromosome 22q11. Tetralogy of Fallot can also be found in association with endocardial cushion defects, most commonly in those fetuses diagnosed with trisomy 21. A variation of tetralogy of Fallot with significant hypoplasia of the conal septum is more commonly found in Asian and Native American populations. In some cases, the distinction between tetralogy of Fallot and double outlet right ventricle can be difficult. Double outlet right ventricle can be classified with tetralogy of Fallot-like physiology if the aorta overrides the ventricular septum more than 50% toward the right ventricle with a lack of fibrous continuity between the aortic and the mitral valves.
Imaging: The anterior malalignment-type VSD is best viewed in the long axis of the heart where the overriding aorta can be easily appreciated (Fig. 16.17). The VSD will not be seen in the four-chamber view as it is located in the outlet conal septum. The short axis views will demonstrate the VSD and the anterior deviation of the conal septum and allow for assessment of right ventricular outflow obstruction. The degree of pulmonary stenosis may progress in utero, and therefore serial imaging should be performed. The pulmonary valve, main pulmonary artery, and branch pulmonary arteries should be carefully measured as they may be hypoplastic (Fig. 16.18). The direction of ductus arteriosus flow is important because with severe obstruction there will be reversed flow and these babies will have significant right to left ventricular-level shunting and will be ductal dependent at birth. There can be varying degrees of abnormalities of the pulmonary valve (see tetralogy of Fallot with pulmonary atresia and tetralogy of flow with absent pulmonary valve).
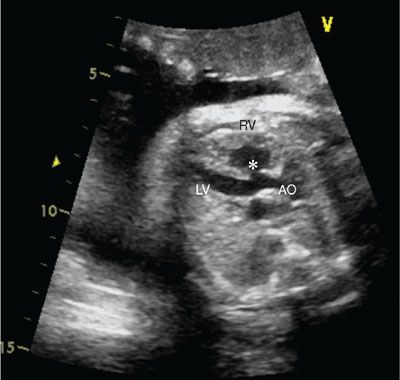
FIGURE 16.17: Long axis view of tetralogy of Fallot with an overriding aorta (AO). The asterisk denotes the ventricular septal defect. RV, right ventricle; LV, left ventricle.
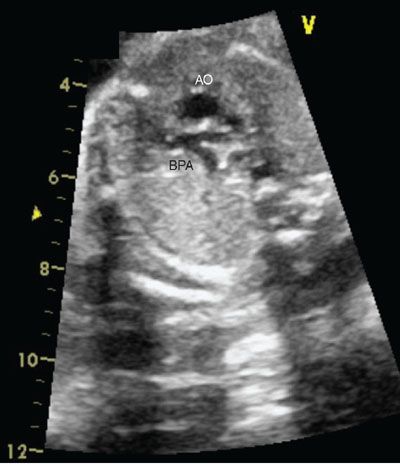
FIGURE 16.18: Three-vessel view in tetralogy of Fallot demonstrating hypoplastic branch pulmonary arteries (BPA) relative to the aorta (AO).
Treatment/Outcome: Surgical repair is necessary in tetralogy of Fallot and usually occurs between 2 and 6 months of age or earlier if cyanosis is present. Surgical repair includes closure of the VSD and relief of the right ventricular outflow tract obstruction. If the pulmonary valve annulus is small, a patch may be placed across the right ventricular outflow tract to relieve valvar obstruction (“transannular patch repair”). If the valve size is within normal limits (>−2 SD), an infundibular patch may be considered. Finally, stenosis or discontinuity of the branch pulmonary arteries, if present, is usually also corrected. Outcomes are good, although some patients may need catheter intervention for residual right ventricular outflow tract or branch pulmonary artery stenosis.
Tetralogy of Fallot with Pulmonary Atresia: Tetralogy of Fallot with pulmonary atresia (TOF/PA) is a severe form of tetralogy of Fallot in which the pulmonary valve is atretic and there is no antegrade pulmonary blood flow. TOF/PA is typically associated with good-sized branch pulmonary arteries if there is a large patent ductus arterious. However, it may be associated with hypoplastic branch pulmonary arteries and aortopulmonary collateral arteries (MAPCAs) arising from the aorta if the ductus is absent. The type and timing of surgical repair of TOF/PA is dependent on the nature of the pulmonary blood supply. Outcome is typically not as good, especially in the presence of hypoplastic pulmonary arteries.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


