Congenital Heart Disease
Congenital heart disease (CHD) occurs in approximately 1% of all live births, or 40,000 births annually in the United States. It remains the leading cause of birth defect–associated death in infancy. Approximately 25% of CHD defects are critical or severe, resulting in symptoms during the first year of life. About 25% of patients with CHD eventually die of their disease, and approximately 25% of CHD-related deaths occur in the first month of life. Survival rates continue to improve. Today 69% to 95% of patients survive to 18 years of age depending on the type of malformation. There is a continually growing population of patients living with corrected CHD, now estimated to be more than 1.5 million individuals in the United States. Patients are living longer due to improved imaging diagnosis as well as medical and surgical treatment. The majority of patients with CHD are diagnosed during fetal assessment or after birth, with echocardiography as the mainstay of initial diagnostic evaluation. Cardiac catheterization has traditionally been the diagnostic gold standard, providing morphologic and functional information including data on pulmonary vascular resistance and oxygen saturation within chambers and vessels. Magnetic resonance imaging (MRI) and computed tomography (CT) now play increasingly important and complementary roles in the delineation of CHD in the preoperative and postoperative periods.
CHD is a complex disease process that occurs during embryologic development of the cardiovascular system. This chapter will review basic cardiac embryology and morphology, and provide an overview of the segmental approach to CHD that is important in understanding and accurately communicating the morphology and physiology of different types of CHD. The classic radiographic findings of CHD that have traditionally been taught to radiologists in training will be reviewed. In addition, this chapter will provide an overview of the anatomy and physiology of common CHDs and emphasize the role of cross-sectional imaging with CT and MRI before and after cardiac surgery.
Embryology
A basic understanding of cardiac embryology is helpful to understand the complex spectrum of abnormalities that may be encountered in CHD. By definition, cardiac embryology reflects the events that occur in the development of the human heart from conception to the eighth week of gestation. The heart, unlike any other organ, must be able to function while it is undergoing embryologic development.
The major events of cardiac development occur between the third and fifth weeks of gestation. The cardiovascular system arises from the mesoderm as the cardiogenic crescent on the 18th day of fetal life. A straight heart tube develops by the 20th day of gestation, and it is at this point that the heart begins to beat. As cells continue to be added to the heart tube, the tube undergoes the looping process. Typically the heart tube loops to the right, which is referred to as D-looping. The heart may also loop to the left, which is referred to as L-looping. The looping of the heart plays a major role in the eventual spatial relationship of the right ventricle (RV) and left ventricle (LV) [ Fig. 3.1 ] .

Once looping is complete during the fourth week of gestation, circulation through the heart is in series from the right atrium (RA) to the left atrium (LA), then the LV to the RV to the truncus arteriosus, which serves as a common outflow tract to the aorta and main pulmonary artery (MPA). During this time, the embryologic aortic arches are also developing into the great arteries and their major branches, as well as the ductus arteriosus. The process of convergence, which aligns the RA with the developing RV and the aorta with the developing LV, follows ventricular looping. The heart then undergoes septation during the fifth week of fetal life. The interventricular septum grows from apex to base to septate the ventricles, and the formation of the septum primum and secundum separates the LA and RA. The endocardial cushion tissue lining the atrioventricular canal (AVC) then septates the common AV valve into separate tricuspid and mitral valves. At the same time, the common outflow tract from the ventricles, or the truncus arteriosus, septates to separate the ascending aorta and the MPA. The infundibulum, or conus of muscle forming the right ventricular outflow tract beneath the pulmonary valve, together with the swirling blood flowing out of the developing ventricles, causes the left and right ventricular outflow tracts and aorta and pulmonary artery (PA) to appear wrapped around one another when they are normally developed (i.e., not parallel in configuration) [ Fig. 3.2 ] . Separation of the right and left heart is nearly complete by the seventh week of fetal life, but does not completely finish until the final closure of the ductus arteriosus and the interatrial septum after birth.

In summary, the major events in cardiac embryology occur between the third and seventh weeks of fetal life, and include the formation of a straight heart tube with common venous inflow to the developing LV and common arterial outflow from the developing RV. This is followed by looping of the tube to the right (D) or to the left (L), differentiation of morphologic chambers with convergence to provide inflow to the RV and outflow to the LV, valve formation, and chamber separation. Abnormal arrest of this process or defects during any stage of development may result in the wide spectrum of CHDs that can be detected at birth.
Identification of Cardiac Chambers and Great Vessels
The first step in the analysis of CHD by cross-sectional imaging is to accurately identify the morphology of the cardiac chambers and vessels. The RA is identified as the chamber that receives the insertion of the inferior vena cava (IVC) and coronary sinus, and also by a broad-based triangular appendage with characteristic pectinate muscle morphology extending to the AV junction. The LA is identified by a narrow-based, tubular, finger-like appendage. The pulmonary vein insertion is not reliable for identification of the LA, because the pulmonary veins are often partially or totally anomalous. The situs of the atria can be solitus (S), inversus (I), or ambiguous (A) [ Fig. 3.3 ].

The RV is identified by the following features:
- 1.
A muscular attachment between the free wall and the interventricular septum called the moderator band
- 2.
An AV valve that is slightly more apical in position than the AV valve of the LV
- 3.
The presence of an infundibulum (or conus), which creates a muscular outflow tract that separates the tricuspid valve and the semilunar valve arising from the RV [ Fig. 3.4 ]

Figure 3.4
Morphologic characteristics of the normal right (RV) and left ventricles (LV).
A, Four-chamber steady-state free precession (SSFP) magnetic resonance image shows the moderator band ( arrow ) of a normal RV. The RV is positioned to the right of and anterior to the LV, and the LV has a smooth superior septal surface. B, Long-axis sagittal oblique SSFP image through the RV profiles the normal tricuspid and pulmonary valves, which are separated by the conus or infundibulum ( arrow ). C, Vertical long-axis SSFP image profiles the normal aortic and mitral valves, which are in fibrous continuity because there is no subaortic conus. Ao, Aorta; LA, left atrium; RA, right atrium.
The LV is identified by its smooth superior septal surface, which is devoid of any muscular attachments, and by the absence of a conus, so that the mitral valve and the semilunar valve arising from the LV are in fibrous continuity.
The PA is recognized as the vessel that supplies branches to the lungs, but not to the body. The aorta is identified as the vessel that supplies the coronary arteries and systemic arteries to the body.
Segmental Approach to Heart Disease
Any combination of atrial, ventricular, and great vessel morphology may occur in CHD. One must take a simple, logical, step-by-step approach to understand and describe disease before initiating treatment. The segmental approach to CHD described by Richard van Praagh [ Fig. 3.5 ] involves determination of the following factors:
- 1.
The morphology of the three major cardiac segments: Viscero-atrial situs, ventricular looping, and the relationship of the great arteries
- 2.
How each segment is connected to the adjacent segment: This determination involves analysis of the AVC, which connects the atria to the ventricles, and evaluation of the ventriculo-arterial connection, including the conus (also called the infundibulum ), which connects the ventricles to the great arteries. Potential AV connections are concordant connection (LA to LV and RA to RV), discordant connection (LA to RV and RA to LV), common atrioventricular canal (CAVC), double-inlet LV, unilateral AV valve atresia, straddling AV valve, and overriding AV valve. The options at the level of the conus are normal connection, transposition of the great arteries (TGA), truncus arteriosus, double-outlet right ventricle (DORV), and rarely, double-outlet LV [ Fig. 3.2 ]. Description of the conal muscle is important, because this muscle may narrow the outflow tracts and influence subsequent surgical management
- 3.
The presence of associated malformations involving the atrial and ventricular septum or the extracardiac vasculature: Examples include but are not limited to the following: ASD, VSD, partial or total anomalous pulmonary venous connection (TAPVC), bilateral superior vena cava (SVC), and coarctation
- 4.
How segmental combinations and connections, along with the associated malformations, function

As an example, using the segmental approach, a patient would be described as “congenitally corrected” {S,L,L} TGA with pulmonary stenosis and a VSD [ Fig. 3.6 ]. Breaking this down to the individual components, there is (1) S-solitus atria, L-looped ventricles, and L-malposed great arteries; (2) a discordant AV connection (LA to RV and RA to LV) and a discordant ventriculo-arterial connection (RV to aorta and LV to PA); (3) pulmonary stenosis and a VSD; and (4) a balanced (physiologically corrected) circulation.

Heterotaxy
Accurate anatomic and physiologic diagnosis of CHD using a structured approach has led to significant improvement in patient outcomes. This method is associated with a more appropriate selection of therapeutic options, whether they be medical, surgical, or both. This is especially true as applied to the patient with complex CHD in the setting of heterotaxy syndrome, or abnormal sidedness of the thoracic and abdominal viscera. With normal development, there is lateralization of the internal thoracic and abdominal organs with respect to the left-right axis, so that the heart is in the left chest with the apex pointing to the left, the stomach and spleen are located in the left upper quadrant, and the liver is located in the right upper quadrant. This is referred to as situs solitus. Situs inversus totalis refers to complete mirror image reversal of the normal position of the thoracic and abdominal viscera and vasculature. Patients with heterotaxy have situs ambiguous or variable lateralization of the thoracic and abdominal viscera and vasculature that does not follow either situs solitus or situs inversus totalis. The exact incidence of heterotaxy is unknown, but it is thought to occur in 1 in 10,000 to 40,000 births, including 3% of all neonates with CHD and 30% who have a cardiac malposition. The prognosis of patients with heterotaxy is primarily based on the severity of the associated cardiovascular malformations.
Heterotaxy encompasses a spectrum of cardiac, vascular, and visceral abnormalities of early embryonic development of the thorax and abdomen that occur in the fifth week of gestation. This spectrum includes abnormalities in septation of the conotruncus, growth of the endocardial cushions, septation between the LA and pulmonary venous plexus, formation of the spleen, rotation of the bowel, and lobation of the lungs. The defining features of heterotaxy therefore include conotruncal anomalies (such as TGA or DORV), endocardial cushion or CAVC defects, anomalous pulmonary and systemic venous connections, symmetry of the atrial appendages with partial or complete absence of the atrial septum, asplenia or polysplenia, malrotation of the bowel, and symmetry of lung lobation (bilateral right- or left-sided morphology).
There is no one feature that is pathognomonic in heterotaxy syndrome. To be able to recognize the numerous different congenital cardiovascular anomalies that may be present, it is helpful to understand that heterotaxy syndrome represents a spectrum between complete bilateral right-sidedness (also called right isomerism or heterotaxy with asplenia ) and bilateral left-sidedness (also called left isomerism or heterotaxy with polysplenia ). Most patients fall somewhere in between these two extremes [ Figs. 3.7 and 3.8 ].


Right isomerism or heterotaxy with asplenia is characterized by bilateral right eparterial bronchi and trilobed lungs, absence of a spleen, a midline transverse liver, and a stomach in variable position. Heterotaxy with asplenia may be associated with complex forms of cyanotic heart disease including complete absence of the atrial septum resulting in a common atrium, CAVC, and DORV and/or pulmonary atresia.
Left isomerism or heterotaxy with polysplenia is associated with bilateral left hyparterial bronchi and bilobed lungs, a midline transverse liver, multiple spleens or splenules that can be located on either side of the abdomen, and a stomach in variable position. Heterotaxy with polysplenia may be associated with intracardiac defects similar to those seen in the setting of asplenia, but they tend to be less severe.
Patients with heterotaxy may have numerous different anomalies of the systemic and pulmonary venous connections, including partial or TAPVC and a bilateral SVC. An interrupted IVC at the level of the liver with azygous continuation is more often seen in polysplenia (bilateral left-sidedness), because the IVC is normally a right-sided structure.
Applying the segmental approach to CHD is crucial in the setting of heterotaxy. This method facilitates accurate characterization of complex and variable morphology, and guides decision making in the best approach for management and surgical repair.
Physiologic Subgroups
Chest radiographs play an important role in evaluating the physiology of heart disease and determining palliative therapy at birth by distinguishing between lesions associated with increased pulmonary blood flow (left-to-right shunts and intermixing states with unobstructed pulmonary blood flow), lesions associated with decreased pulmonary blood flow due to right-sided obstruction, and lesions associated with pulmonary venous hypertension due to systemic (left) sided obstruction [ Fig. 3.9 ]. Intermixing states are usually due to the presence of a large shunt in lesions associated with a common atrial or single ventricular chamber. Clinically, cyanosis is present due to tremendous mixing of deoxygenated blood with oxygenated blood before it enters into the systemic circulation.

With the current diagnostic accuracy of echocardiography for CHD, chest radiography now plays a lesser role in diagnosing the morphology of CHD. In the past, chest radiographs were analyzed together with physiologic information gleaned from clinical examination (murmurs, cyanosis, etc.) to construct a differential diagnosis that would determine the initial treatment needed by the patient at birth. Radiographic classification of CHD may be based on pulmonary blood flow as follows:
- I.
Increased pulmonary vascularity
- A.
Central left-to-right shunts (acyanotic)
- 1.
Ventricular septal defect (VSD)
- 2.
Atrial septal defect (ASD)
- 3.
Atrioventricular septal defect/common atrioventricular canal (CAVC)
- 4.
Patent ductus arteriosus (PDA)
- 5.
Less common lesions: partial anomalous pulmonary venous connection (PAPVC), aortopulmonary window, coronary-cameral fistula
- 1.
- B.
Peripheral left-to-right shunts
- 1.
Vein of Galen malformation
- 2.
Hepatic hemangioendothelioma
- 3.
Large extremity arteriovenous malformations
- 1.
- C.
Hyperdynamic states, such as thyrotoxicosis and anemia
- D.
Intermixing states without obstruction to pulmonary blood flow (cyanotic)
- 1.
Transposition of the great arteries (TGA)
- 2.
Truncus arteriosus
- 3.
Double outlet right ventricle (DORV)
- 4.
Single ventricle
- 5.
TAPVC (unobstructed)
- 1.
- A.
- II.
Decreased pulmonary vascularity
- A.
Tetralogy of Fallot (TOF)
- B.
Pulmonary atresia with VSD
- C.
Intermixing states with obstructed pulmonary blood flow (cyanotic)
- 1.
TGA with pulmonary stenosis
- 2.
Truncus arteriosus with pulmonary stenosis
- 3.
DORV with pulmonary stenosis/atresia
- 1.
- A.
- III.
Pulmonary venous congestion and edema
- A.
Left-sided obstructive lesions
- 1.
Cor triatriatum
- 2.
Hypoplastic left heart syndrome (HLHS)
- 3.
Shone syndrome
- 4.
Bicuspid aortic valve and aortic stenosis
- 5.
Aortic coarctation
- 1.
- B.
Hyperdynamic states
- C.
Severe left-to-right shunts
- A.
- IV.
Normal pulmonary vascularity
- A.
Mild left-sided obstructive lesions
- B.
Intermixing states with balanced pulmonary blood flow
- C.
Mild right-sided obstruction—pulmonary valve stenosis
- A.
Treatment of Congenital Heart Disease
Immediate Palliation
The need for immediate palliative therapy of CHD at birth depends on the patient’s clinical and hemodynamic status. The type of palliation is determined by echocardiographic and radiographic findings. For example, in a patient with increased pulmonary vascularity, PA banding to diminish pulmonary blood flow may be considered. In a patient with decreased pulmonary vascularity caused by outflow tract obstruction, pulmonary blood flow can be augmented by surgical relief of the obstruction or placement of a systemic arterial-to-pulmonary arterial shunt such as a modified Blalock-Taussig shunt (BTS) [ Fig. 3.10 ]. Older adults with CHD may have had the classic BTS or Waterston or Potts shunts, which are no longer routinely used. Intermixing states are treated in the neonatal period to balance and optimize pulmonary and systemic blood flow. Pulmonary venous hypertension caused by left-sided obstruction is treated through relief of the cause of obstruction.

Permanent Palliation
After immediate palliation, decisions regarding permanent palliation are based on the number of functioning ventricles and the feasibility of separating deoxygenated and oxygenated blood flows. When there are two well-formed, normal-sized, functioning ventricles in conditions such as TOF, TGA, truncus arteriosus, and some forms of CAVC and DORV that are amenable to a two-ventricle repair, the anatomy is rearranged to restore physiologic venous return and unobstructed systemic and pulmonary arterial outflows.
When there is only one normal-sized, well-functioning RV or LV, this ventricle will be used as the systemic ventricle, and the pulmonary circulation will be palliated by establishing direct connections between the systemic veins and pulmonary arteries or cavopulmonary connections. This is called single-ventricle repair [ Fig. 3.11 ]. Conditions that can be palliated with single-ventricle repair include unbalanced CAVC, tricuspid atresia, HLHS, double-inlet LV, DORV, and pulmonary atresia with an intact ventricular septum. Occasionally, conditions with two functioning ventricles may end up in the single-ventricle pathway because of an inability to separate the deoxygenated and oxygenated circulations. This may occur with some forms of DORV, straddling AV valves, and crisscross AV valves. A list of named surgical procedures is provided in the appendix to this chapter (see section “Named” Pediatric Cardiac Surgical Procedures ).

Indications for Imaging by Computed Tomography and Magnetic Resonance Imaging
In the preoperative period, CT and MRI are used to evaluate CHD when complete characterization of pathology by echocardiography is inadequate due to suboptimal acoustic windows. Evaluation by echocardiography may be suboptimal with abnormalities involving the extracardiac vasculature, such as aortic coarctation, anomalous pulmonary venous connection, systemic venous anomalies, branch PA stenosis, and anomalous coronary artery origin.
In the postoperative period, the goals of imaging change considerably. Information about cardiac function and flow and potential postoperative complications is needed in addition to morphologic assessment. The goals of postoperative imaging are to evaluate ventricular and valvular function, survey the patency of surgically created grafts, conduits, and baffles, and gather information that will aid in the timing of necessary surgical or catheter-based reintervention. Typical indications for postoperative imaging that will help guide subsequent management include the evaluation for recurrent coarctation, the determination of timing of catheter-based or surgical pulmonary valve replacement in repaired TOF, the investigation of the cause of cyanosis after the Fontan operation, the evaluation of the systemic RV after intraatrial repair of TGA, and screening evaluation before staged single-ventricle repair. The role of MRI and CT increases in the postoperative period and in older patients when acoustic windows diminish.
Common Conditions
Left-to-Right Shunts
Communications between the systemic and pulmonary circuits that do not result in cyanosis include intracardiac left-to-right shunts and large systemic arteriovenous connections. Common examples of the former are VSD, ASD, AVC defect, and PDA. Less common examples are partial anomalous pulmonary venous connection, aortopulmonary window, and coronary-cameral fistula. Examples of peripheral left-to-right shunts are infantile hepatic hemangioendotheliomas and large systemic arteriovenous malformations such as the vein of Galen malformation in the brain and Parkes-Weber syndrome affecting the extremities.
Ventricular Septal Defect
After bicuspid aortic valve, VSD is the second most common congenital intracardiac lesion in children, accounting for 20% to 30% of cases of CHD. Five percent of VSDs are associated with chromosomal abnormalities such as trisomies 13, 18, and 21. Many small VSDs close spontaneously. The classification of VSDs on the basis of location [ Fig. 3.12 ] and size is important in deciding whether medical (expectant) or surgical treatment is indicated.
Muscular VSDs are defects in the muscular septum [ Fig. 3.13 ]. They may be classified according to their location in the septum (midmuscular, apical muscular). “Swiss cheese septum” describes multiple muscular VSDs.
An AVC-type VSD is a defect in the AVC portion of the septum [ Fig. 3.13 ]. Also known as an inlet VSD, this condition is commonly seen in the setting of heterotaxy.
Several different terms are used for outflow tract VSDs or defects in the interventricular septum associated with the outlet or subaortic or subpulmonary region of the heart. Conoventricular VSDs are a form of outlet VSD occurring within or at the junction between the conal septum and the muscular septum. Conoventricular VSDs may be in close proximity to the semilunar valves, and are also referred to as subarterial VSDs. Clinically, it is important to evaluate for associated aortic valve prolapse and aortic regurgitation.
The strictest definition of a membranous VSD is a defect of only the membranous septum, which would result in a relatively small defect. In day-to-day use, the term perimembranous or paramembranous VSD usually implies that the defect includes the region of the membranous septum, but also extends into the muscular septum, conal or outflow tract septum, or AVC septum. Outflow tract VSDs can be associated with malalignment, in which the conal septum of the RV outflow tract is displaced. This may create a substrate for outflow tract obstruction. Anterior malalignment of the conal septum often results in the RV outflow obstruction seen in TOF (described later), and posterior malalignment can lead to a VSD with LV outflow tract obstruction and an arch abnormality such as coarctation or complete arch interruption.
A VSD classically presents with a prominent, blowing, pansystolic murmur heard best at the left lower sternal border. Affected patients may have congestive heart failure (CHF), repeated respiratory infections, or failure to thrive. Presentation is usually after the first month of life when pulmonary vascular resistance normalizes and the degree of shunting increases. In an older child, a large, untreated VSD may lead to elevated pulmonary vascular resistance and pulmonary hypertension, which can resolve if the shunt is closed. In rare cases, pulmonary vascular obstructive disease or irreversible pulmonary hypertension can develop, and may even cause reversal of blood flow. This leads to a right-to-left shunt across the septal defect (formerly known as “Eisenmenger physiology”), and eventually to cyanosis. In most cases, corrective surgery is performed before flow reversal across the shunt occurs.
Conventional chest radiograph findings of a left-to-right shunt are usually evident when the ratio of pulmonary to systemic blood flow is greater than 2 : 1. Pulmonary vascularity is increased, and there is dilatation of the pulmonary arteries and cardiomegaly [ Fig. 3.13 ]. A VSD generally results in volume loading of the LA and LV, which may appear prominent on chest radiographs. When pulmonary vascular resistance normalizes after the first month of life, pulmonary blood flow may further increase, resulting in radiographic signs of pulmonary vascular congestion and edema.
Up to 50% of VSDs close spontaneously. Those that do not may be treated with surgical closure of the defect. VSDs that are difficult to reach surgically can be treated with device closure at cardiac catheterization.
Atrial Septal Defect
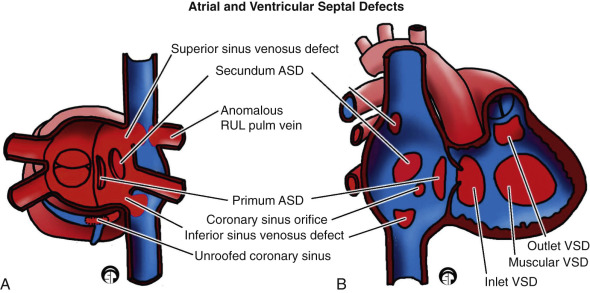
ASD is the second most common cardiac anomaly in children, accounting for 10% of all cases of CHD. An ASD is the most common intracardiac shunt that persists into adulthood. It is found six to eight times more commonly in females than in males. ASDs are classified according to location in the atrial septum as follows [ Fig. 3.12 ] :
- •
A patent foramen ovale [ Fig. 3.14 ] represents defective apposition of the septum secundum and the septum primum, which normally fuse together after birth.

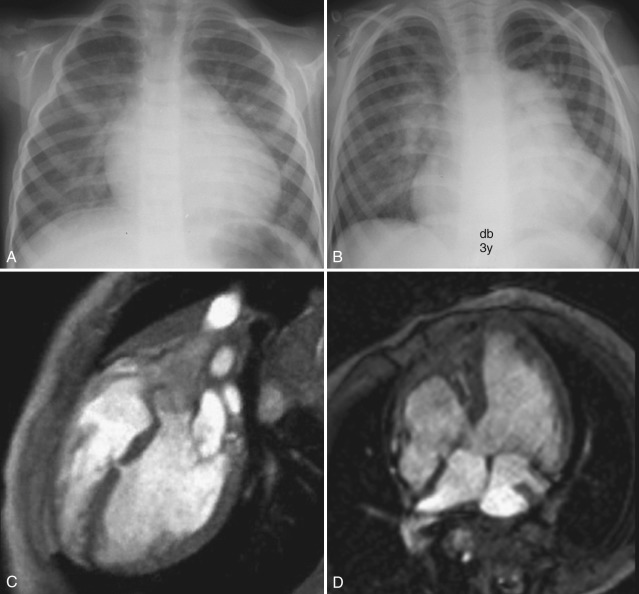
Figure 3.14
Types of atrial septal defects (ASDs).
A, Four-chamber steady-state free precession (SSFP) image shows a large secundum ASD ( asterisk ) that is shunting left to right, resulting in dilation of the right atrium ( RA ) and right ventricle ( RV ). B, Sagittal oblique SSFP image through the interatrial septum shows the flap of a patent foramen ovale ( arrow ).Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree