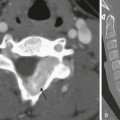
Chapter 114 Overview, Etiology, and Clinical Presentation: A multicystic dysplastic kidney (MCDK) is the most common form of cystic renal dysplasia. With high-grade obstruction or atresia of the upper urinary tract (renal pelvis and/or ureter) during early renogenesis, disordered parenchymal development results in an MCDK.1 Two types of MCDK exist: the pelvoinfundibular type (common) and the hydronephrotic type (uncommon).2,3 MCDK can occur in half of a duplex kidney (usually the upper moiety) or in renal fusion anomalies, such as a horseshoe kidney and crossed fused renal ectopia. MCDK is sporadic in most cases, although familial cases have been described.1,4 Vesicoureteral reflux (VUR) (both ipsilateral and contralateral) and contralateral ureteropelvic junction obstruction are common.1,5 Long-term sequelae (e.g., infection and hypertension) are rare, because most affected kidneys either partially or completely involute on their own over time.6–9 Imaging: On ultrasound, computed tomography (CT), and magnetic resonance imaging (MRI), a classic MCDK is characterized by multiple cysts of various sizes (ranging from 1 mm to several cm) that do not communicate, with no normal intervening renal parenchyma (Fig. 114-1). The affected kidney may be small, normal, or enlarged.1 Compensatory hypertrophy of the contralateral kidney often is present.10 The hydronephrotic form has cysts that communicate, mimicking pelvocaliectasis, but no normal functioning renal tissue.2 The diagnosis of both classic and hydronephrotic MCDK can be confirmed by diuretic renal scintigraphy (e-Fig. 114-2) or MR urography (MRU) (e-Fig. 114-3). A voiding cystourethrogram (VCUG) may reveal VUR into a blind-ending ureter on the side of the MCDK.11,12 Figure 114-1 A multicystic dysplastic kidney in a 3-month-old boy. e-Figure 114-2 A multicystic dysplastic kidney in a 6 month-old boy. e-Figure 114-3 A multicystic dysplastic kidney and magnetic resonance (MR) imaging, hydronephrotic form. Treatment: Follow-up ultrasound typically demonstrates involution of the cysts over time, eventually resulting in remnant dysplastic renal tissue that may be impossible to visualize.13,14 Immediate resection of an MCDK is rarely indicated15 and is reserved for cases in which the size of the kidney is a problem. In the past, routine nephrectomies were performed because of a fear of malignant degeneration. Current belief discounts this possibility, and as long as no other complication exists, surgery is not indicated. Overview and Etiology: Autosomal-dominant polycystic kidney disease (ADPKD), the most common form of inherited renal cystic disease, is seen in 1 in 800 live births.16,17 Two types of ADPKD exist, each with clearly defined chromosomal mutations. The more severe form (PKD1) accounts for about 85% to 90% of cases, whereas the milder phenotype (PKD2) presents later in life, results in less morbidity, and is less common.16,18 Cysts arise from both the renal cortex and the medulla.16,19 Clinical Presentation: Systemic hypertension, hematuria, and slowly progressive renal insufficiency are the typical clinical manifestations.1,20 ADPKD is very rarely detected prenatally because fewer than 5% of nephrons are cystic before birth.1 Diagnosis of ADPKD early in life portends a poor prognosis; 43% of affected persons die in the first year of life, hypertension develops in 67%, and some affected children progress rapidly to end-stage renal disease.20 Because it can be difficult to delineate between autosomal-recessive and autosomal-dominant forms of polycystic kidney disease in a neonate, screening of siblings and parents may be helpful. Imaging: The diagnosis of ADPKD usually is made on the basis of ultrasound (Fig. 114-4), although CT or MRI also can suggest it. Although the kidneys may be normal during the first decade of life, they also may contain one or more simple cysts (Fig. 114-5).16,21 The kidneys may be abnormally enlarged and echogenic, mimicking autosomal-recessive polycystic kidney disease (ARPKD) in early life (e-Fig. 114-6).16,22 Although it is typically a bilateral process, imaging findings may be asymmetric, and in young children they can manifest as a unilateral abnormality.16,21,23 Microscopic cysts, as well as larger cysts that can be detected by cross-sectional imaging techniques, are common in the liver and less frequent in the pancreas, spleen, lungs, thyroid, ovaries, and testes.19 The remainder of the urinary tract is usually normal. Figure 114-4 Autosomal-dominant polycystic kidney disease in a newborn. Figure 114-5 Autosomal-dominant polycystic kidney disease in an 8-year-old girl. e-Figure 114-6 Autosomal-dominant polycystic kidney disease (ADPKD) in a neonate mimicking autosomal-recessive polycystic kidney disease (ARPKD). Treatment: Although renal function is generally maintained during childhood, ADPKD is responsible for the need for chronic dialysis in 10% to 12% of adult patients.1 Hypertension should be managed medically to prevent long-term complications. About 12% to 15% of patients with ADPKD have an intracranial aneurysm, most often arising from the circle of Willis.19,23–25 Screening MR angiography usually is not performed in childhood, however, because aneurysm rupture is rare in this period. Overview: ARPKD is a rare disorder (occurring in 1 : 20,000 live births) that affects both kidneys.26,27 Genetic studies have localized the inheritance of ARPKD to chromosome 6p.27,28 The PKHD1 gene is expressed in fetal and adult kidneys and livers, the two major sites affected by the disease.28 Approximately 1 in 70 persons are a carrier for this condition.26,27 Clinical Presentation: ARPKD has been subdivided into categories based on clinical presentation. The perinatal and neonatal forms, which have the poorest prognosis, are associated with severe renal dysfunction and minimal hepatic portal tract fibrosis. Affected children often die of the disease early in life.27,29 Infantile and juvenile forms are associated with less severe renal dysfunction and more significant hepatic fibrosis.27 The degrees of renal and hepatic abnormalities tend to be inversely proportional.17,27 A number of children with ARPKD also have Caroli disease (see Chapter 88).27 Pathology: Histologically, the kidneys have a spongelike appearance, replaced by myriad minute 1- to 2-mm cysts of relatively uniform size, representing markedly dilated and elongated collecting ducts.1,27 These cysts radiate from the hilum to the surface of the kidney without a clear demarcation between the medulla and cortex. Fibrosis is present in the renal interstitium. Children with significant hepatic involvement are at increased risk for complications related to portal hypertension, and ascending cholangitis can lead to sepsis and death.27 Imaging: Abdominal radiography in the neonatal period commonly reveals bilateral abdominal or flank masses with centralized gas-filled bowel loops. The lungs may appear small because of a combination of pulmonary hypoplasia and mass effect from marked nephromegaly (e-Fig. 114-7).27 e-Figure 114-7 Autosomal-recessive polycystic kidney disease in a newborn boy. On ultrasound, affected neonatal kidneys tend to be smoothly enlarged (frequently more than four standard deviations above the mean expected length for age), are hyperechoic as a result of the multiple acoustic interfaces at the walls of the dilated ducts, and have poor corticomedullary differentiation (e-Fig. 114-8).1,27,29,30 On occasion, discrete cysts (solitary or multiple) of variable size (most commonly <1 cm) may be seen (Fig. 114-9).26,30 A sonolucent rim may be seen at the periphery of the kidney, especially in older infants, as a result of cortical compression and relative sparing.26,27 Contrast-enhanced CT (Fig. 114-10) or MRI show delayed opacification of the renal parenchyma with a prolonged and striated nephrogram. When studied with MRI, the dilated tubules in affected kidneys appear linear and hyperintense on T2-weighted imaging, radiating from the medulla toward the cortex (Fig. 114-11).31 Figure 114-9 Autosomal-recessive polycystic kidney disease in a 6-month-old boy presenting with acute renal failure and bilateral renal masses. Figure 114-10 Autosomal-recessive polycystic kidney disease. Figure 114-11 Autosomal-recessive polycystic disease. e-Figure 114-8 Autosomal-recessive polycystic kidney disease in a newborn girl. Treatment: Eighty-six percent of patients who survive beyond 1 month of age are alive at 1 year, and 67% are alive at 15 years.32 In infants who have initially adequate (although not normal) renal function, the disease may not be detected until later in the first decade, when ultrasound is being performed for unrelated purposes. Systemic renovascular hypertension may occur and require medical management.17,27 Persons with ARPKD who have severely impaired renal function eventually will require renal replacement therapy, either dialysis or renal transplantation. Many rare inherited syndromes and genetic disorders are associated with renal cystic disease (Box 114-1). Overview, Etiology, and Clinical Presentation: A calyceal diverticulum represents a focal outpouching of the renal collecting system that is located within the renal parenchyma and that contains urine.33 This condition has an incidence of 3.3-4.5 : 1000 in children, based on excretory urography.34–36 Calyceal diverticula are likely developmental in origin; they are lined with urothelium and surrounded by muscularis mucosa.33,35 Although these structures most commonly are an incidental finding, they can be associated with both infection and calculus formation as a result of urinary stasis.33,35,37 Imaging: Abdominal radiographs may be normal or may demonstrate one or more calculi (or milk of calcium) within a calyceal diverticulum.35 Upon ultrasound, such diverticula may mimic a solitary renal cyst, appearing as a round, thin-walled anechoic structure (Fig. 114-12).34 They can vary in size ranging from a few millimeters to several centimeters (with a mean size around 11 mm).35,37 They most commonly occur within the upper pole of the kidney.33 Upon excretory phase imaging (including excretory urography, MRI, and CT), calyceal diverticula typically fill with contrast material (Fig. 114-13). A thin connection, or neck, to an adjacent calyceal fornix may or may not be identified.35 Figure 114-12 Calyceal diverticulum. Figure 114-13 Calyceal diverticulum in a 4-year-old girl. Overview, Etiology, and Clinical Presentation: Congenital infundibulopelvic stenosis is a very rare congenital anomaly of the renal collecting system that presents as unilateral or bilateral caliectasis. Although the exact etiology of this condition is uncertain, Lucaya et al.39 hypothesized that this abnormality is a milder manifestation of the process that results in multicystic dysplastic kidney. Affected children may be predisposed to urinary tract infections, and this condition may be associated with other renal and urinary tract anomalies.39 Imaging: Upon contrast-enhanced excretory phase imaging studies (including excretory urography, MRU, and CT), this condition has a specific appearance: the renal collecting system infundibula are abnormally narrowed but not entirely obliterated, and the calyces appear abnormally rounded and dilated (Fig. 114-14). The renal pelvis also appears narrowed as a result of stenosis or hypoplasia. This condition may be unilateral or bilateral. Despite the cystlike appearance of the calyces on such contrast-enhanced studies, the ultrasound appearance of the kidneys can be normal. Renal function may be normal to severely impaired.39 Figure 114-14 Infundibulopelvic stenosis simulating cystic kidney disease. Congenital Megacalyces (Megacalycosis) Overview, Etiology, and Clinical Presentation: Congenital megacalyces is a very rare anomaly of the renal collecting system that may be easily confused for hydronephrosis.40 Calyceal involvement may be unilateral or bilateral. This abnormal calyceal dilatation is not due to urinary tract obstruction but instead is likely a result of underdevelopment/hypoplasia of the renal medullary pyramids.41,42 Boys are more commonly affected than are girls. Associated urinary stasis may predispose affected children to urinary tract infection and urolithiasis.41 Imaging: Congenital megacalyces may be misidentified as obstructive caliectasis by ultrasound. With excretory urography, however, this condition is easily identifiable because of the presence of abnormally dilated calyces that lack normal papillary impressions, have a polygonal or faceted shape, and appear to be increased in number (20 or more calyces may be seen) (Fig. 114-15).42 The renal parenchyma overlying affected calyces may appear thin. The renal pelvis can be normal in caliber or mildly dilated,41 and classically, no related urinary tract obstruction should be present. This appearance of the calyces on MRU and excretory-phase CT (CT urography) also should suggest the diagnosis. Figure 114-15 Congenital megacalyces in a 6-year-old boy. Overview: Ureteropelvic junction (UPJ) obstruction is the most common type of congenital urinary tract obstruction, occurring in 3 per 1000 live births. Boys are more frequently affected than are girls, and the left kidney is more commonly involved than the right kidney. About 30% of cases are bilateral, although severity of obstruction and pelvocaliectasis may be asymmetric.1 Pathophysiology: An intrinsic narrowing (e.g., smooth muscle deficiency, increased fibrosis and collagen, stricture, valve, kinking, and altered peristalsis) occurs at the level of the UPJ, leading to dilatation of the pelvicalyceal system.1,43 On occasion, extrinsic narrowing can be seen from a crossing renal vessel in older children and adults.44 UPJ obstruction often is seen in association with other urinary tract anomalies, such as contralateral multicystic dysplastic kidney, upper urinary tract duplication,45,46 VUR,47 and ureterovesical junction obstruction.48 Clinical Presentation: When UPJ obstruction is not detected prenatally, affected children commonly present with a palpable abdominal mass in the neonatal period. Older children may present with intermittent hydronephrosis and flank pain, often associated with crossing vessels. UPJ obstruction can be complicated by calculi, infection (pyonephrosis), and hemorrhage, especially when diagnosis is delayed.29 Imaging: On ultrasound, abnormal dilatation of the pelvicalyceal system of varying degrees is seen, whereas the ureter is normal in caliber.1,29 Ultrasound findings suggestive of renal dysplasia may be present in cases of severe obstruction (described later). Debris within the collecting system can represent infection or hemorrhage (Fig. 114-16). Careful interrogation of the UPJ region with Doppler ultrasound may identify a crossing vessel, when present.49 Frequently, separating dilated nonobstructed from dilated obstructed renal collecting systems is difficult by ultrasound alone. Figure 114-16
Congenital and Neonatal Abnormalities
Kidney and Ureter
A longitudinal ultrasound image of an enlarged left kidney shows multiple noncommunicating simple cysts of variable size, mimicking hydronephrosis. The very small amount of remaining renal parenchyma is abnormally echogenic.
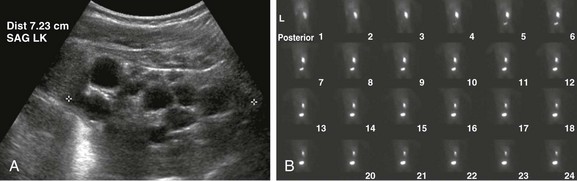
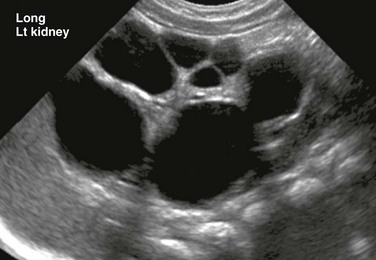
A, A longitudinal ultrasound image of the left kidney shows multiple noncommunicating cysts of variable size. Visualized renal parenchyma is abnormally echogenic. B, Posterior diuretic renal scintigraphy images show no appreciable left kidney function with no uptake or excretion of radiotracer. Right kidney function is normal.
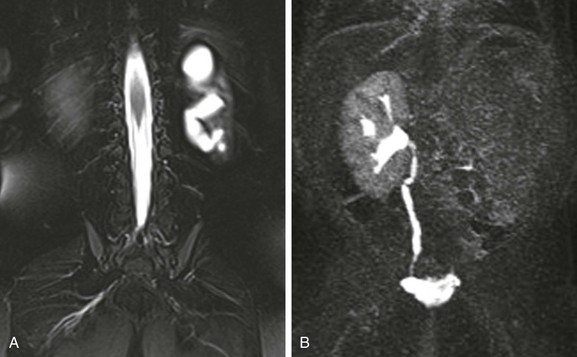
A, A coronal T2-weighted MR image with fat saturation shows a fluid signal within the left kidney that has a configuration similar to that of the renal collecting system. Only a small amount of left kidney parenchyma is present. B, A coronal T1-weighted MR urogram maximum intensity projection image obtained 15 minutes after contrast material injection confirms the absence of the left kidney function. The right kidney is normal.
Autosomal-Dominant Polycystic Kidney Disease
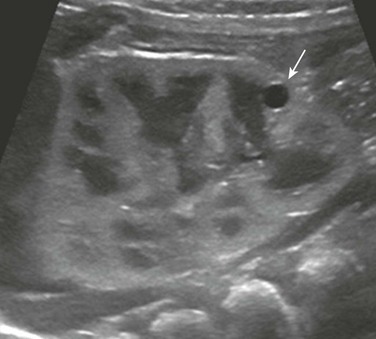
A longitudinal ultrasound image shows a small lower pole simple renal cyst (arrow). The renal cortex appears echogenic as well. Three additional simple renal cysts were identified in this neonate with a maternal history of autosomal-dominant polycystic kidney disease.
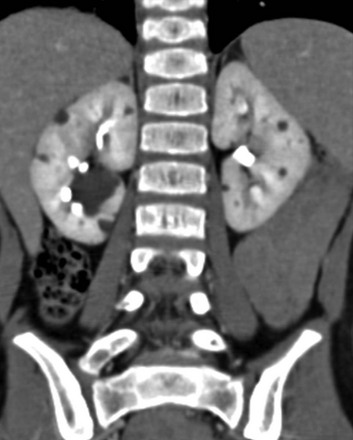
A coronal reformatted contrast-enhanced computed tomography image shows multiple low attenuation lesions in both kidneys due to the presence of cysts.
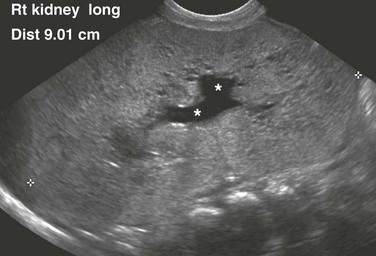
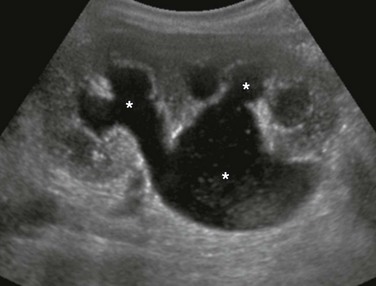
A longitudinal ultrasound image shows a markedly enlarged echogenic kidney with loss of corticomedullary differentiation. Multiple tiny cystic spaces are present within the kidney. Mild pelvocaliectasis (asterisks) is present as well. Although the imaging features favor ARPKD, a maternal history of ADPKD led to disease.
Autosomal-Recessive Polycystic Kidney Disease
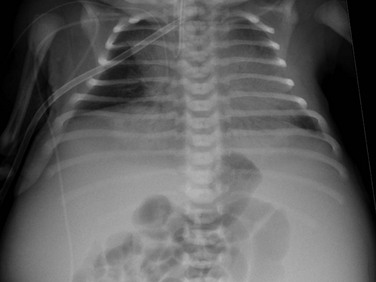
An anteroposterior chest radiograph shows small lungs due to pulmonary hypoplasia. Bulging of the flanks and centralization of bowel gas is due to bilateral nephromegaly.
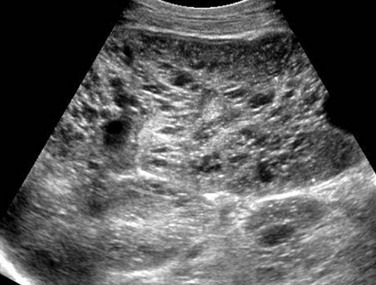
A longitudinal ultrasound image of the right kidney shows nephromegaly, loss of normal corticomedullary differentiation, and innumerable tiny cystic structures replacing the renal parenchyma.
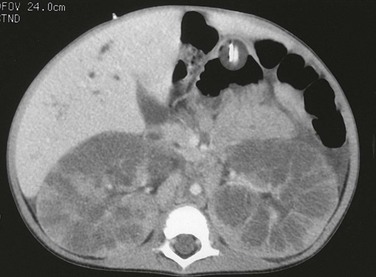
A computed tomography image shows that the pathologic process is primarily medullary. The dilated tubules have produced a linear pattern that can be seen clearly in a few regions. Marked cortical thinning present along the anterior aspect of each kidney is not a typical finding, because the cortex is usually spared.
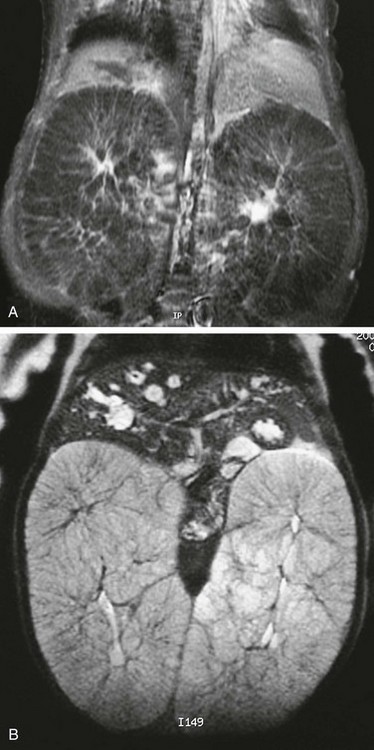
T1-weighted (A) and T2-weighted (B) magnetic resonance images of an infant with autosomal-recessive polycystic disease. The kidneys are enormously enlarged, and on the T1-weighted image, the spoke wheel is appreciated. The patient also has Caroli disease as seen by the dilated biliary system on the T2-weighted image.
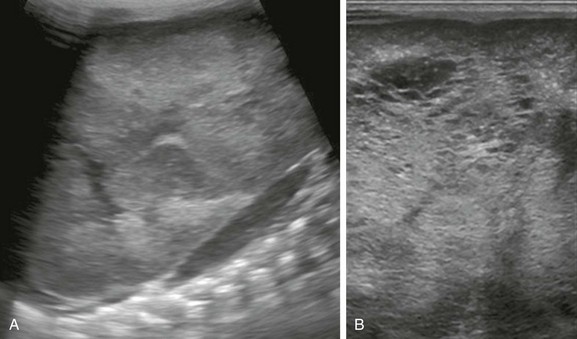
A, A longitudinal ultrasound image of the left kidney confirms the presence of a nephromegaly with loss of corticomedullary differentiation. B, Further assessment of the kidney using a linear high-frequency transducer reveals numerous tiny cystic structures within the kidney due to dilated ducts and tubules. The peripheral cortex is relatively hypoechoic.
Other Renal Cystic Diseases in Children
Calyceal Diverticulum
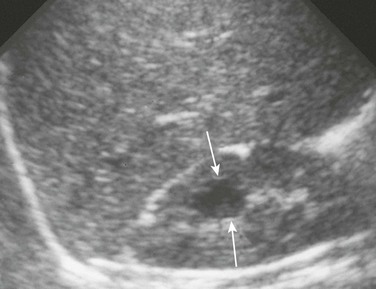
A longitudinal ultrasound image reveals a medullary cyst (arrows). On a subsequent intravenous urography procedure, the cyst filled with contrast material.
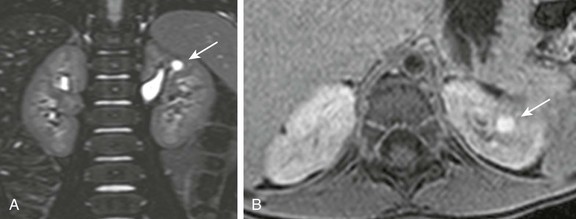
A, A coronal three-dimensional T2-weighted fast spin echo magnetic resonance (MR) image with fat saturation shows a round structure within the upper pole of the left kidney demonstrating signal intensity identical to urine. B, An axial delayed postcontrast T1-weighted MR image with fat saturation reveals excreted contrast material filling this abnormality.
Congenital Infundibulopelvic Stenosis
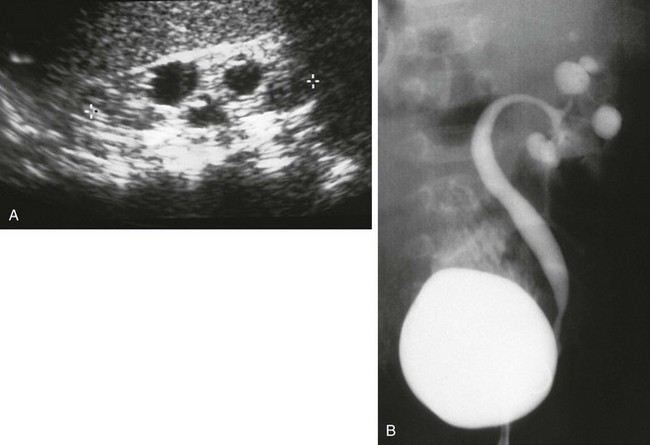
A, A longitudinal ultrasound image shows multiple “cysts” scattered throughout the kidney in this child with VACTERL association. B, A voiding cystourethrogram shows vesicoureteral reflux into the abnormal pelvocalyceal system. The ureter has a greater caliber than the renal pelvis, infundibula, and calyces. The most peripheral portion of each calyx is rounded, producing the cystic spaces seen with ultrasound.
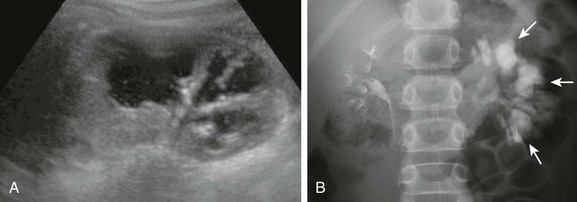
A, A longitudinal ultrasound image shows multiple dilated calyces with overlying parenchymal thinning. Debris within the renal collecting system was due to infection. B, A subsequent excretory urography image demonstrates contrast material filling an increased number of left kidney polygon-shaped calyces (arrows), confirming the diagnosis of congenital megacalyces.
Ureteropelvic Junction Obstruction
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree