Chapter 106 Overview: The colon is the least involved segment in intestinal atresias; colonic atresia constitutes between 1.8% and 15% of intestinal atresias, with an overall incidence of approximately 1 in 20,000 births. Colonic stenosis is very rare, with fewer than 15 cases reported in the literature. It typically consists of stricturelike stenosis, although membranous stenosis also has been reported. The anatomic descriptive classification of small intestinal atresias (see Chapter 103) also is applied to colonic atresias.1,2 Colonic atresia is associated with other colonic abnormalities, as well as extracolonic and extraintestinal abnormalities. Approximately 22% of patients also may have Hirschsprung disease or hypoganglionosis.3,4 Extraintestinal anomalies include the musculoskeletal system, heart, abdominal wall, eyes, and central nervous system.5 Etiology: The etiology of colonic atresia is thought to be similar to that of small bowel atresias, that is, related to an ischemic event in utero that leads to resorption of the involved bowel and discontinuity of the proximal and distal segments.6 More recently, mutations involving fibroblast growth factor 10 or its receptor have resulted in intestinal atresia in mice, which suggests that genetic determinants also may play a role.7 Clinical Presentation: Infants with colonic atresia present with abdominal distension, vomiting, and failure to pass meconium. Polyhydramnios is uncommon, because the atresia is sufficiently distal to allow resorption of swallowed amniotic fluid. The onset of vomiting may be delayed compared with more proximal atresias and may become feculent.5,8 Patients with colonic stenosis demonstrate similar findings proportionate to the degree of stenosis. Imaging: Abdominal radiographs in patients with colonic atresia show distal obstruction with multiple dilated loops of bowel and abdominal distension with flank protuberance and diaphragmatic elevation. However, the proximal colon characteristically is dilated far more than the other loops of bowel (Fig. 106-1, A). This disproportionate dilatation is due to competence of the ileocecal valve, which allows intestinal contents forward into the proximal colonic segment but does not allow decompression proximally. Air-fluid levels are seen on horizontal beam radiographs, and prone images show no gas in the rectum (Fig. 106-1, B). Figure 106-1 Colonic atresia. A contrast enema performed on these patients demonstrates a microcolon that terminates blindly, devoid of the filling defects characteristic of a meconium ileus (Fig. 106-1, C). Treatment: The treatment of colonic atresia is surgical. Because 15% to 20% of patients with colonic atresia also have proximal intestinal atresia, the proximal bowel should be evaluated. Hirschsprung disease should be excluded with suction biopsy before reestablishing intestinal continuity, and coexisting extraintestinal anomalies should be identified.5 Overview: Anorectal malformations encompass the spectrum of anal atresia and stenosis, with an incidence of approximately 1 : 5000 live births worldwide. Approximately one third of patients have isolated lesions, but two thirds of patients are affected by other abnormalities encompassing the gastrointestinal tract and multiple other systems. Approximately 95% of patients have a fistula to the urethra (males), vagina (females), or the perineum; however, 95% of patients with Down syndrome and anorectal malformation have no fistula.9 Anatomically, anorectal malformations are classified as high, intermediate, or low lesions, depending on whether the atresia lies above or below the levator sling. Some authors object to this classification because the anomaly is a spectrum, rather than three separate and distinct types. The classification system of Levitt and Peña (Table 106-1) provides both prognostic information and implications for optimal surgical management.9 These authors note that girls with high lesions with a high rectovaginal fistula who later present with a persistent/unrepaired urogenital sinus abnormality actually represent girls with a persistent cloaca, which are classified by the length of the persistent cloacal canal.10 Table 106-1 Classification of Anorectal Malformations From Levitt M, Peña A. Anorectal malformations. In: Coran A, et al. eds. Pediatric surgery. Philadelphia, Mosby; 2012. Rectal atresia often is discussed among the anorectal malformations, although patients with rectal atresia have a normal anal canal and external physical findings. The rectum is atretic 1 to 2 cm above the anus.10 Etiology: Anorectal atresia with rectourethral or rectovaginal fistulas may be the result of failure of the urorectal septum to descend to the cloacal membrane and the eventual site of the perineal body; if the cloaca is too small, the hindgut may then terminate anteriorly, entering either the urethra in the male or the vagina in the female. Rectoanal atresias are thought to be related to vascular accidents, similar to the atresias that occur in the small bowel and colon. Imperforate anus is due to failure of breakdown of the anal membrane.11 No single genetic abnormality is associated with anorectal malformations. The most common chromosomal abnormalities are trisomy 21 and a microdeletion at chromosome 22q11.2, although abnormalities in multiple chromosomes have been identified.12 Approximately 15% of patients with rectovestibular or rectoperineal fistulas, which are indicative of low lesions, have a positive family history for anorectal malformations. Genetic studies in both animals and humans have implicated defects in the sonic hedgehog, Wnt5a, and Skt genes. These studies suggest that the pathogenetic mechanisms of high and low fistulas differ and that fistula formation may be due to a genetic mutation rather than obstruction. Clinical Manifestations: Anorectal malformations are clinically apparent at birth, with the exception of patients with rectal atresia, who have a normal external appearace. Physical examination may reveal a perineal or vestibular fistula, although the possibility of such a fistula is best evaluated after 24 hours, because meconium may not appear in the perineum before this time. If fecal material appears in the urine, a rectourethral fistula may be inferred. Patients who demonstrate a “flat bottom” (i.e., the buttock crease is not visible) have poor development of pelvic musculature and a more guarded prognosis.9 Girls with a single perineal orifice have a cloacal malformation (Fig. 106-2). Figure 106-2 Physical examination findings in boys and girls with anorectal malformations. The best known group of anomalies associated with anorectal malformations is the VACTERL association (vertebral anomalies, anal atresia, cardiac abnormalities, esophageal atresia with or without tracheoesophageal fistula, and renal and limb abnormalities). Associated genetic syndromes include trisomy 21, trisomy 8, and fragile X syndrome.12 Cardiovascular abnormalities are seen in about one third of patients, most commonly atrial septal defect and persistent ductus arteriosus, followed by tetralogy of Fallot and ventricular septal defect.9 Vertebral anomalies occur in approximately one third of patients (Fig. 106-3, A and B), and their severity may be correlated with the complexity of the anorectal lesion. Coronal clefts often are seen in patients with imperforate anus and have been reported as being nine times more common in boys than in girls.13 Esophageal atresia with tracheoesophageal fistula is seen in approximately 10% of patients, and abnormalities affecting the duodenum (atresia or malrotation) are found in 1% to 2%. Hirschsprung disease, although reported in these patients, is rare.14 Genitourinary anomalies are common, presenting in one third to one half of cases, again correlated with increasing complexity of the defect. These anomalies range from reflux to renal dysplasia or agenesis, with cryptorchidism and hypospadias seen in male patients. Females may demonstrate müllerian abnormalities, including duplications and obstructions, particularly in the setting of cloacal abnormalities.9,10 Figure 106-3 High anorectal malformation in a male neonate. Imaging: The goal of imaging is to elucidate associated anomalies and to determine fistulous anatomy. The sacral ratio9 quantifies the degree of sacral hypoplasia by measuring the distance between the iliac crests and the inferior border of the sacroiliac joints (A-B) and the distance between the inferior border of the iliac joints and the tip of the sacrum (C-D); the CD/AB ratio should be >0.77. An increase in the ratio to approach 1.0 indicates better sacral development and better prognosis. Several imaging methods have been used for evaluation of the atretic anatomy in patients without a perineal fistula. The invertogram relies on visualizing the gas-filled rectum and relating its termination to pelvic bony landmarks—particularly extension below the coccyx, which would classify it as a low lesion.9 However, problems such as meconium distal to gas and movement of the rectum with infant straining and crying often render this examination inexact and problematic. Voiding cystourethrography can be useful in visualizing the fistula if contrast material flows retrograde into the atretic distal colon during voiding (Fig. 106-3, C). Voiding cystourethrography does not always work because meconium may obstruct the fistula. Instilling water-soluble contrast material into the rectal pouch after a colostomy often is successful in outlining fistulas to the bladder (Fig. 106-3, D) or urethra (Fig. 106-3, E). Occasionally, gas may outline some of the pertinent anatomy (e-Fig. 106-4). e-Figure 106-4 High anorectal malformation in a female neonate with a cloaca. Ultrasound also is used to evaluate the distal position of the rectum (Fig. 106-5). The meconium in the pouch allows easy visualization of the distal rectum; however, changes in apparent position with straining make it somewhat challenging. Measurements of 10 mm ± 4 correlate with a low lesion, whereas measurements of 24 ± 6 tend to correlate with intermediate and high lesions.15 Ultrasound can identify the puborectalis musculature; the presence of this musculature correlates with low-type lesions, whereas its absence correlates with high-type lesions.16 Magnetic resonance imaging (MRI) also has been used to identify the internal anatomy. High field strengths, small field of view, and sequences without fat suppression, which increase the conspicuity of the muscles of the pelvic floor, are helpful in visualizing the position of the distal rectal pouch with respect to the levator mechanism.17 MRI also can be used to evaluate the rectum and levator sling after surgery (Fig. 106-6). Figure 106-6 Postoperative magnetic resonance after anal atresia repair. Patients with rectal atresia who have normal external anatomy are evaluated with contrast enema. The enema demonstrates a very short distal rectum that terminates blindly (e-Fig. 106-7). e-Figure 106-7 Rectal atresia. Treatment: The treatment of anorectal malformations is surgical but varies with the level and complexity of the lesion. Most boys can undergo repair with a posterior sagittal approach alone, although some with a high lesion also will require an abdominal approach to mobilize a very high rectum. In girls, 30% of cloacas need to be repaired through an abdominal approach. Babies with rectal atresia typically undergo an initial colostomy, with later anastomotic repair of the atretic rectum with the anal canal.10 Overview: Although Currarino syndrome initially was described in three infants in 1981 as a triad of anorectal malformation (anorectal stenosis) with rectoperineal fistula,18 sacral anomaly (classically crescentic abnormality resembling a scimitar), and a presacral mass,19 this abnormality actually encompasses a much wider spectrum of phenotypic abnormalities and underlying genetic defects. Etiology: Currarino syndrome is an autosomal-dominant disorder with variable penetrance; approximately 50% of the cases are sporadic. A mutation located at 7q36 encoding for the HLXB9 gene responsible for nuclear transcription factor HB9 plays a role in the dorsoventral separation of the caudal embryo; other genes related to the same differentiation pathway also may exist.20,21 Clinical Presentation: The patients typically present with constipation and may undergo anorectal dilatation. The presacral mass is often small and may not be detected unless the syndrome is suspected; an unrecognized presacral mass may present later with complications such as meningitis or development of malignancy.20–22 The spectrum of presacral masses includes teratoma, rectal duplication cyst, anterior meningocele, leiomyosarcoma, and ectopic nephroblastoma. Other abnormalities that can be seen in the full spectrum of the syndrome include Hirschsprung disease, dysganglionosis, bladder motility abnormalities, vesicoureteral reflux, rib anomalies, urinary and gynecologic abnormalities, cord tethering and intraspinal lipomas.18,23 Imaging: Radiographs of the sacrum may reveal the typical scimitar-shaped sacral abnormality (Fig. 106-8, A); other sacral defects, typically deletion abnormalities, may be present. In a minority of patients the sacrum is normal.21 Figure 106-8 Currarino syndrome. Those patients with anorectal stenosis may have a rectoperineal fistula or an anteriorly placed anus and present with constipation, sometimes in early adulthood. Contrast enema shows marked narrowing of the distal rectum, with excessive stool proximally (Fig. 106-8, B). If the presacral mass is of sufficient size, it will exert a visible mass effect upon the posterior wall of the distal rectum. MRI best demonstrates the presacral masses well and provides a window into the spinal canal (Fig. 106-8, C). In young infants, the presacral mass often is visible sonographically (Fig. 106-8, D). Overview: Hirschsprung disease is the result of failure of normal bowel innervation as a result of the arrest of proximal to distal migration of vagal neural crest cells, and therefore it is considered a neurocristopathy. As a result of abnormal arrest of migration, a variable length of distal bowel lacks parasympathetic Auerbach (intermuscular) and Meissner (submucosal) plexuses and is unable to participate in normal peristalsis, resulting in failure of relaxation and functional obstruction. Pathologic evaluation also shows abnormal acetylcholinesterase staining and hypertrophied nerve fibers (e-Fig. 106-9). e-Figure 106-9 Pathologic findings in Hirschsprung disease. Hirschsprung disease occurs in approximately 1 per 5000 live births and is responsible for approximately 15% to 20% of cases of neonatal bowel obstruction, presenting in the newborn period in approximately 80% of cases. The incidence is less among African Americans and Asian Americans, quoted as 2.1 and 2.8 per 10,000 live births, respectively.24 The transition point between normal and abnormal bowel can occur anywhere and can extend through a variable length of the small bowel; rarely, total intestinal aganglionosis is present.24 The presence of the transition point at the rectosigmoid is termed short-segment aganglionosis and occurs in approximately 80% to 90% of cases. Males predominate in the incidence of short-segment aganglionosis (the boy : girl ratio is 4 : 1); however, male preponderance diminishes in longer segment involvement. Hirschsprung disease is most commonly associated with Down syndrome, which is present in approximately 2% to 10% of patients with Hirschsprung disease, with a 5 : 1 boy to girl ratio.24 Other syndromes associated with Hirschsprung disease include Waardenburg, Shprintzen-Goldberg, McKusick-Kaufman, Bardet-Biedl, Currarino syndrome, and with central hypoventilation (Ondine syndrome), termed Haddad syndrome.24–26 Patients with longer segment aganglionosis are more likely to demonstrate Haddad syndrome than are those with short-segment disease.27 Between 5% and 30% of patients with Hirschsprung disease have limb, skin, central nervous system, kidney, cardiac, and other malformations.25 Etiology: The etiology of Hirschsprung disease is not precisely known. One theory is that the migrating ganglion cells do not reach the distal segment because they are fewer in number or mature prematurely. However, it also is possible that the ganglion cells reach the distal bowel but perish or are unable to proliferate because of a deficient microenvironment.28 Multiple genetic mutations have been identified, with the most common being the RET proto-oncogene located at 10q11, which is found in approximately 15% to 35% of sporadic cases and 50% of familial cases. EDNRB, which is located at 13q22, is identified in approximately 5% of cases.29,30 A positive family history is encountered in a minority of patients (<10%), but this percentage increases to nearly 25% in patients with total aganglionosis, with the degree of risk proportional to the length of the affected segment and the degree of consanguinity.31,32 Clinical Presentation: Hirschsprung disease in the neonate presents with distal obstruction, including abdominal distension and bilious vomiting. Failure to pass meconium beyond the first 24 hours is seen in up to 90% of patients.28 Rarely, perforation of the cecum or appendix can occur in neonates, typically in those with long-segment disease.33,34 In older children, Hirschsprung disease presents with constipation, abdominal distension, and vomiting, with failure to thrive in more severe cases. In late presentations, the zone of transition is typically low.28
Congenital and Neonatal Disorders
Colonic Atresia and Stenosis
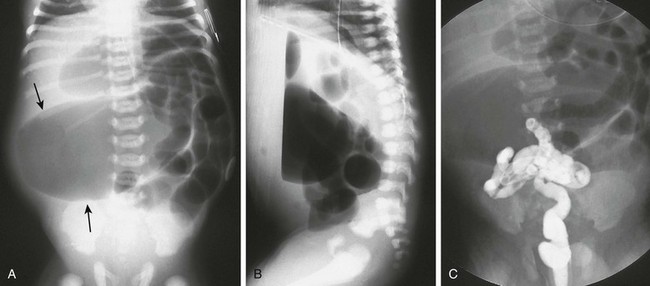
A, A supine abdominal radiograph of a neonate with abdominal distension and bilious vomiting demonstrates protuberance of the flanks and elevation of the diaphragms. Gas-filled dilated loops of bowel are present, but one loop is dilated far more than other loops, and represents the atretic proximal colonic segment (arrows). An orogastric tube decompresses the stomach. B, A prone cross-table lateral radiograph shows a large air-fluid level located within the atretic proximal colonic segment. No gas is present in the rectum. C, A contrast enema shows a microcolon terminating blindly at the atretic segment.
Anorectal Malformations
Males
Females
Perineal fistula
Perineal fistula
Rectourethral fistula
Vestibular fistula
Bulbar
Persistent cloaca
Prostatic
≤3 cm common channel
Recto-bladder neck fistula
>3 cm common channel
Imperforate anus without fistula
Imperforate anus without fistula
Rectal atresia
Rectal atresia
Complex defects
Complex defects
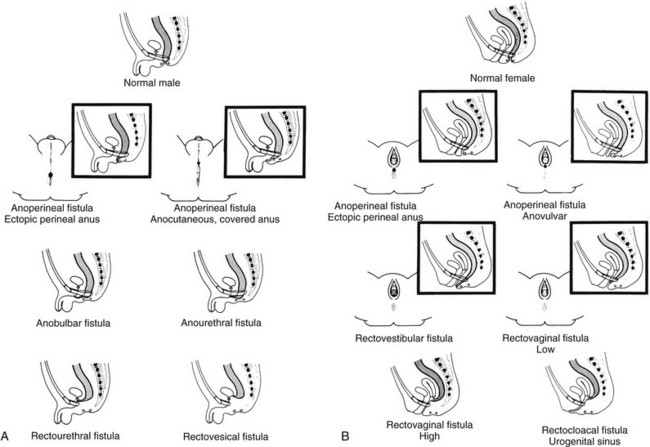
A, A schematic diagram of normal and ectopic termination of the hindgut in the male. B, A schematic diagram of normal termination and common sites of ectopic termination of the hindgut in the female. (From Santulli TV. In: Mustard WT, et al, eds. Pediatric surgery. ed 2. St Louis: Mosby–Year Book; 1969.)
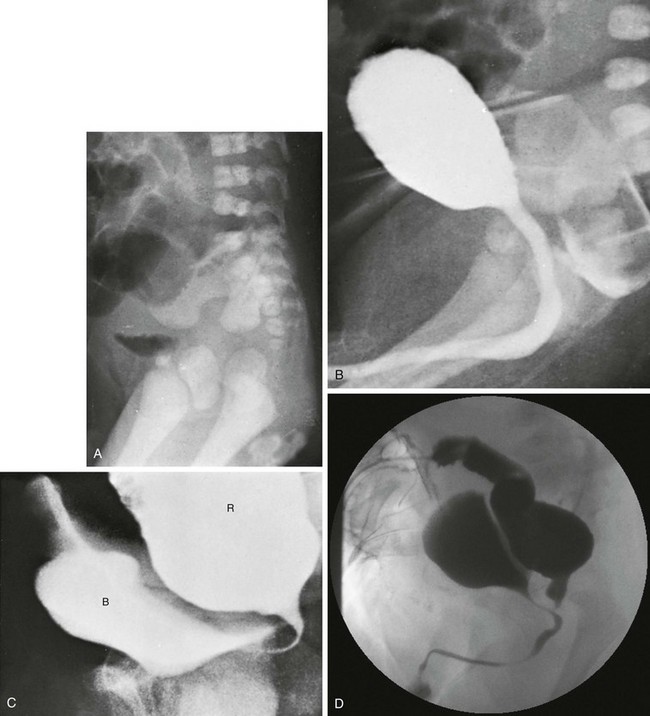
A, A lateral radiograph of the pelvis and lower abdomen the demonstrating gas in the urinary bladder. In addition, numerous lumbar coronal clefts are identified. B, A voiding cystogram demonstrates a rectoprostatic fistula. Note how the rectum bulges inferiorly below the level of the fistula. This accounts for the spuriously small distance sometimes seen between rectal gas and anal marker in such patients, although the bowel edge may lie above the puborectalis sling. C, Examination of the rectal pouch with contrast material identifies a rectovesical fistula in a male infant. D, Examination of the rectal pouch with contrast material shows the fistulous connection to the posterior urethra in a male infant. (A, From Berdon WE, et al. The radiologic evaluation of imperforate anus. An approach correlated with current surgical concepts. Radiology. 1968;90(3):466-471.)
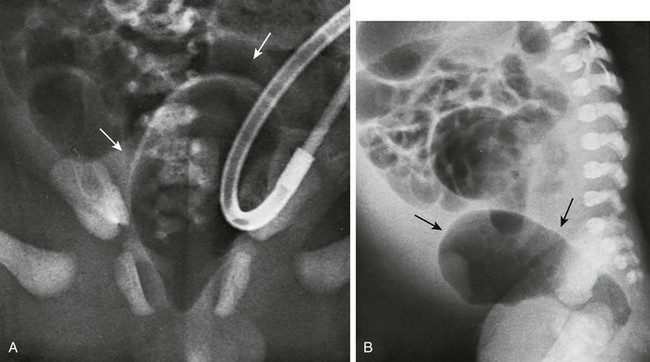
A, A pneumovagina (arrows) represents gas entering a giant vagina (arrows) above the narrow urogenital sinus. Urine also fills the vagina because of the coexisting urogenital sinus. B, A pneumovagina (arrows) in an infant with a rectovaginal and urethrovaginal connection. (From Berdon WE, et al. The radiologic evaluation of imperforate anus. An approach correlated with current surgical concepts. Radiology. 1968;90(3):466-471.)
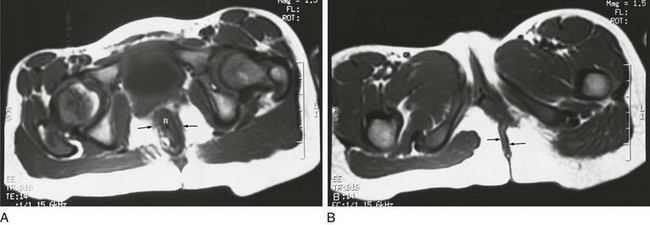
A, An axial T1-weighted section at the level of the pubic bone shows the rectum (R) contained within the puborectalis muscle (arrows). B, An axial T1-weighted image through the region of the pubic bone on another child after surgery shows that the rectum (R) is not enclosed by muscle on the left side. Little puborectalis muscle is seen on the left.
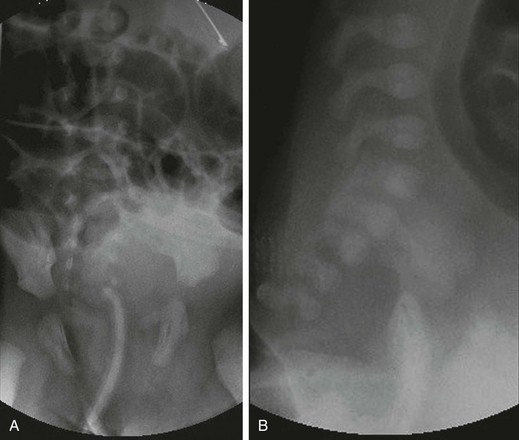
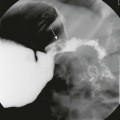
A, An anteroposterior view of a neonate with distal bowel obstruction during an attempt to perform a contrast enema. The catheter is in place but would not advance further. Note the multiple dilated loops of bowel and the decompressing orogastric tube in the left upper quadrant. B, A lateral view during same procedure shows the inability of contrast material to progress further than the tip of the catheter, instead spilling onto the fluoroscopy table.
Currarino Association
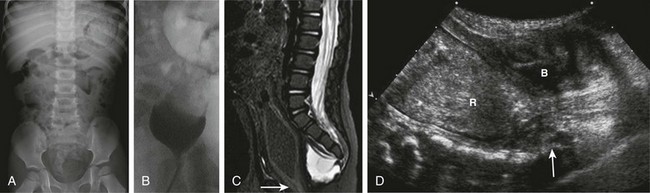
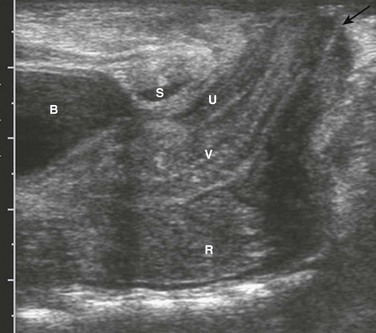
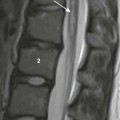
A, A supine abdominal radiograph in 23-month-old boy with a history of constipation demonstrates an increased amount of stool in the colon and a deletion anomaly along the left side of the sacrum, resembling a scimitar. B, A contrast enema demonstrates narrowing of the distal rectum. C, A sagittal T2-weighted lumbar spine image demonstrates a cystic retrorectal mass that proved to represent an anterior meningocele. Note the narrowing of the distal rectum (arrow). D, An anterior sagittal pelvic sonogram of a different male neonate shows a dilated proximal rectum (R) distally narrowed and a retrorectal mass (arrow) that proved to represent a mature teratoma. B, Bladder.
Hirschsprung Disease
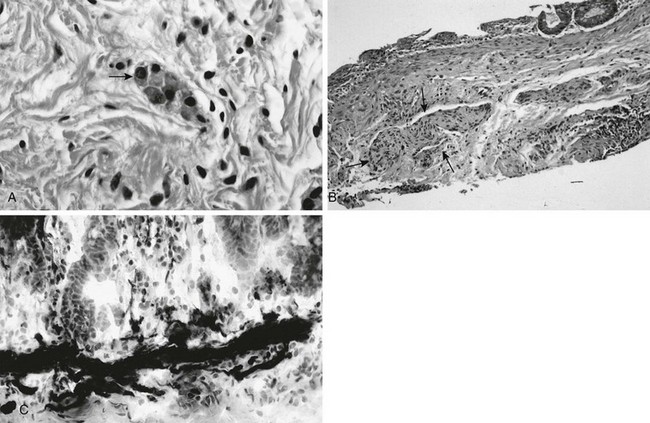
A, In a normal newborn, a suction biopsy and hematoxylin and eosin staining reveals submucosal ganglion cells (arrow). B, Hirschsprung disease, suction biopsy shows submucosal, abnormally thick nerves (arrows) and no ganglion cells. C, Hirschsprung disease, acetylcholinesterase stain of a biopsy specimen reveals abnormal stain of twiglike nerve processes in the lamina propria. (Courtesy Dr. R. Raba, Children’s Hospital of Michigan, Detroit, MI.)
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree