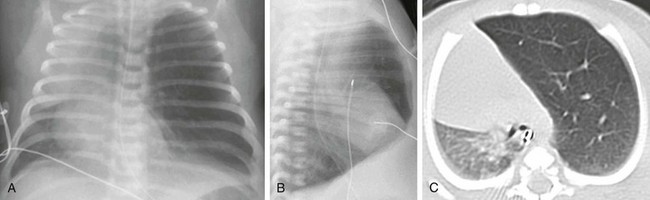
Chapter 53 Congenital lung anomalies refer to a heterogeneous group of pulmonary developmental disorders that affect the lung parenchyma, the arterial supply, and the venous drainage to the lung, or a combination of these entities. The reported incidence of congenital lung anomalies ranges from 1.2 : 10,000 to 1 : 35,000 pregnancies; however, these reports may be an underestimate of their true incidence.1–3 Although prenatal sonography (ultrasound), advances in postnatal imaging, and, more recently, fetal magnetic resonance imaging (MRI) have enhanced our understanding of congenital lung anomalies, substantial controversy continues regarding the nomenclature, classification, pathogenesis, description, and management of these lesions. In addition, considerable variability exists in their prenatal clinical presentation and outcome, ranging from in-utero involution to severe hydrops and fetal demise. Likewise, their postnatal clinical presentation also is variable, ranging from the completely asymptomatic newborn to the older child or young adult with recurrent pneumonias. In this chapter, the underlying etiology, clinical presentation, imaging findings, and management of various congenital lung anomalies encountered in the pediatric population are discussed. The classification of congenital lung anomalies is challenging and continuously controversial from embryologic, radiologic, pathologic, and clinical viewpoints. Several classifications and terminologies with their own advantages and disadvantages have been suggested.4–6 Some investigators have used embryology as a basis and have classified congenital lung anomalies according to the stage of intrauterine development in which the insult resulting in the malformation developed.4 Other investigators have categorized lesions based on their morphologic-radiologic features and divided them into two groups: whole lung malformations (e.g., lung hypoplasia) and focal malformations (e.g., bronchial atresia).5,7 Recently the Langston6 classification has become one of the most accepted classification systems for congenital lung anomalies, particularly from the pathological standpoint, although it is by no means the most widely used classification by all clinical groups. Langston categorizes the wide spectrum of respiratory tract malformations primarily as bronchial atresia, congenital pulmonary airway malformation (CPAM), extralobar bronchopulmonary sequestration (BPS), congenital lobar hyperinflation (CLH), and bronchogenic cysts. These five congenital lung anomalies comprise approximately 90% of the anomalies seen in clinical practice. However, this classification system is limited because other congenital lung anomalies (e.g., pulmonary arteriovenous malformation [AVM]) are not included. For the purpose of relatively clear classification, easy differentiation on imaging studies, and preoperative assessment of surgical lesions, congenital lung anomalies discussed in this chapter are categorized according to their morphologic-radiologic-pathologic features. Such a classification system views congenital lung anomalies as a continuum ranging from predominantly parenchymal abnormalities (i.e., abnormal lung parenchyma, relatively normal vasculature, airway, and foregut derivatives, e.g., CPAM), to predominantly vascular abnormalities (i.e., normal lung parenchyma, normal airway, and no foregut abnormality but abnormal vasculature; e.g., AVM), to combined parenchymal and vascular abnormalities (e.g., pulmonary sequestration and scimitar syndrome) in which influencing factors (foregut and airway components) play an important role and the major abnormalities are intertwined (Fig. 53-1). Figure 53-1 Diagram of the spectrum of congenital lung anomalies, including foregut and airway components. Given that specific terminology for these lesions may be controversial and occasionally confusing, we and other authors8–11 recommend that radiologists thoroughly describe all imaging findings of congenital lung anomalies rather than try to categorize the lesions by pathologic terminology. Specific imaging findings of congenital lung anomalies that need to be evaluated and described include (1) location of lesions, (2) associated anomalous vascular supply and drainage of the lesions, (3) internal components and the degree of aeration, (4) exclusion of communication with the gastrointestinal (GI) tract, (5) integrity of the diaphragm, and (6) an assessment of associated anomalies, such as vertebral anomalies.8,10 Etiology: Bronchial atresia refers to atresia of a lobar, segmental, or subsegmental bronchus at or near its origin resulting in a blind-ended atretic proximal bronchus. Bronchial atresia most frequently affects a segmental bronchus. The precise etiology of bronchial atresia remains unknown, but etiologies such as a vascular insult to the involved atretic or stenotic portion have been proposed.6,8 Several authors who used modern dissecting techniques found that bronchial atresia is more common than originally thought.6,12,13 Furthermore, bronchial atresia and BPSs coexist in nearly all cases,6,12,13 whereas it is found in nearly 70% of CPAM lesions.12 A malformation sequence resulting from airway obstruction during development has been proposed as the unifying element for such a wide spectrum of imaging appearances. Differences in degree, level, and timing of the bronchial obstruction are thought to be responsible for the association of bronchial atresia and other congenital lung anomalies.6,8,12 Bronchial atresia usually is diagnosed as an incidental finding on chest radiographs later in life in asymptomatic older children or adults.6,10 However, bronchial atresia increasingly is being diagnosed in utero, given the widespread use of prenatal imaging.6,9,10,12,13 Imaging: Prenatally, the involved portion of the lung appears hyperexpanded and shows increased homogenous echogenicity on ultrasound and high T2 signal on fetal MRI14 (Fig. 53-2). On occasion, it is possible to identify the centrally located, mucus-filled bronchocele/mucocele on prenatal ultrasound or MRI9,15 (e-Fig. 53-3). Figure 53-2 Bronchial atresia. e-Figure 53-3 Bronchial atresia. Characteristically, the apicoposterior segmental bronchus of the left upper lobe is most commonly affected, followed by the segmental bronchi of the right upper, right middle, and lower lobes.16 In children, radiographic and computed tomography (CT) imaging features of bronchial atresia are characterized by a tubular or glove-shaped opacity representing mucus plugging in the region distal to the atretic bronchus, surrounding segmental hyperlucency due to air trapping, and decreased underlying vascularity15,17 (e-Fig. 53-4). However, in neonates, a portion of the lung distal to the atretic segment may remain atelectatic as a result of the in-utero mucostasis. Therefore some authors recommend avoiding immediate postnatal imaging evaluation because the under-aerated lung related to bronchial atresia may be difficult to differentiate from the normal lung with expected fetal fluid retention in the newborn period.10 Identification of the hallmark atretic bronchus and the “bronchocele/mucocele” by means of two-dimensional (2D) multiplanar or three-dimensional (3D) reconstructions either on prenatal or postnatal imaging may be helpful.10,18 e-Figure 53-4 Bronchial atresia in an older child. Treatment and Follow-up: Management of bronchial atresia is somewhat varied. In general, surgical resection is primarily recommended in symptomatic pediatric patients because of recurrent infection.17,19 Although opinions vary, some centers advocate elective surgical resection of bronchial atresia even in asymptomatic pediatric patients because of potential future lung infection and increased association with CPAM.13,20 Etiology: Bronchogenic cysts result from abnormal tracheobronchial branching and presumably originate from an aberrant bud of the developing foregut, similar to other foregut duplication cysts. Bronchogenic cysts typically are unilocular, fluid-filled, or mucus-filled cysts lined by respiratory epithelium and are attached to but do not communicate with the tracheobronchial tree.6,17,19 Although most bronchogenic cysts are located within the mediastinum (predominantly near the carina), they may be encountered anywhere from the suprasternal area to the retroperitoneum. Bronchogenic cysts also may be found within the lung parenchyma, usually in the lower lobes.17,19 Such intrapulmonary bronchogenic cysts do not communicate with the airway unless superimposed infection with wall necrosis occurs,6 which may further predispose them to recurrent infections. The clinical symptomatology of affected pediatric patients primarily depends on the mass effect the lesion exerts on its neighboring structures including the airway, GI tract, and cardiovascular structures. Airway compression is usually mild, but it may be life threatening in some instances when large bronchogenic cysts are located near the carina regions.21 Imaging: A bronchogenic cyst typically presents as a round or oval-shaped cystic lesion located near the right paratracheal or subcarinal area within the middle mediastinum. Bronchogenic cysts are anechoic on prenatal ultrasound and show high signal intensity on T2-weighted prenatal MRI imaging14,19 (e-Fig. 53-5). On chest radiographs, a bronchogenic cyst manifests as a well-delineated round or oval-shaped middle mediastinal mass. On CT, approximately 50% of bronchogenic cysts demonstrate fluid attenuation value (~0 Hounsfield unit) (Fig. 53-6). The remaining bronchogenic cysts may have CT attenuation higher than water because of thick mucoid, milk-of-calcium, proteinaceous, or hemorrhagic contents. MRI, which can confirm the cystic nature of the bronchogenic cysts on T2-weighted images, is helpful for differentiating bronchogenic cysts with high attenuation value from a mildly enhancing solid mass on CT. Typically, no internal contrast enhancement is seen within the uncomplicated bronchogenic cysts on CT or MRI. The presence of an air-fluid level, thick wall enhancement, or surrounding inflammatory changes often is associated with superimposed infection.11,16,17 Figure 53-6 A bronchogenic cyst in a 15-month-old child with stridor. e-Figure 53-5 Bronchogenic cyst. Treatment and Follow-up: Complete surgical resection is the current management of choice for bronchogenic cysts, particularly in symptomatic pediatric patients.22 Temporizing or palliative procedures such as transparietal, transbronchial, or mediastinal aspiration may be considered in symptomatic pediatric patients who are not surgical candidates. Etiology: CLH, also referred to as infantile lobar emphysema or congenital lobar emphysema, presumably is caused by an intrinsic or extrinsic bronchial narrowing, resulting in subsequent air trapping. Intrinsic bronchial narrowing can be caused by weakness or absence of underlying bronchial cartilage, whereas extrinsic bronchial narrowing may be due to the compression from adjacent mediastinal masses or enlarged vessels. CLH clinically presents with respiratory distress in the newborn period14,17,19 in nearly half of the cases and by the age of 6 months in 80% of cases.23 There is a slight male predominance, and the upper lobes are affected more frequently than the lower lobes, with the left lung affected more often than the right.23 Imaging: On prenatal imaging, CLH manifests as a homogeneously hyperechogenic lesion on ultrasound or as a T2 hyperintense lesion on MRI without visible cysts.9,24 On prenatal imaging, CLH often is indistinguishable from other congenital lung anomalies, particularly bronchial atresia.9,24 CLH usually is diagnosed by its typical clinical presentation and characteristic radiographic features of progressive lobar hyperexpansion and hyperlucency, producing displacement or compression of adjacent structures. In the immediate postnatal period, CLH initially may appear as an area of increased opacity related to retained fetal lung fluid, which will clear on subsequent studies.6,17,19,23 Similar imaging findings are noted on CT, and the attenuated pulmonary vasculature is a helpful clue to distinguish CLH from a pneumothorax or other entities in cases of inconclusive chest radiographic findings17 (Fig. 53-7). Etiology: CPAMs, formerly known as cystic adenomatoid malformations of the lung, were first described in the literature by Ch’In and Tang in 1949 as rare lung lesions occurring in premature or stillborn infants with significant hydrops.26,27 CPAMs are characterized by a heterogeneous group of congenital cystic and noncystic lung masses that communicate with an abnormal bronchial tree lacking supporting cartilage.8,17,28 In 1977, Stocker et al.28 classified these lesions based on their clinical and pathologic features, with subdivisions based on the size of the cysts (types I, II, and III) and according to the location of suspected development of the malformation along the airway. Type I CPAMs consist of cysts larger than 2 cm, with presumed bronchial/bronchiolar origin. Type II CPAMs consist of cysts smaller than 2 cm, with presumed bronchiolar origin. Type III CPAMs appear solid, with a presumed bronchiolar/alveolar origin. However, Stocker later expanded his CPAM classification into five types that included type 0 CPAMs, with presumed tracheobronchial origin, and type IV CPAMs, with presumed distal acinar origin. The term CPAM was now implemented instead of cystic adenomatoid malformations, because cystic changes were observed in only three of the aforementioned types (types, I, II, and IV), and adenomatoid change was observed only in type III.9,17,29,30 Increasing evidence indicates that type IV CPAM lesions and type I pleuropulmonary blastomas may represent the same entity.6,31 It is important to recognize that although Stocker’s classification is widely used, it is by no means universally accepted. For an example, Langston6 classifies CPAM lesions into two types and terms the Stocker type I CPAM as “large cyst type lesions” and the Stocker type II CPAMs as “small cyst type lesions” based on cyst size and pathologic criteria.6 Langston proposed that the type III CPAM actually represents a form of pulmonary hyperplasia and should be excluded from the CPAM classification. Imaging: Prenatally, CPAMs are classified on the basis of cyst size as microcysts (<5 mm) and macrocysts (≥5 mm) on fetal ultrasound and MRI.32 Type 1 CPAMs may contain one, several, or multiple macrocysts, some of which are ≥5 mm in diameter9 (Fig. 53-8). Type II CPAMs have variable appearances, ranging from homogeneously hyperechoic or hyperintense lesions to microcystic lesions exhibiting multiple, uniform cysts that measure <5 mm9,14,19 (e-Figs. 53-9 and 53-10). Figure 53-8 Congenital pulmonary airway malformation type I. e-Figure 53-9 Congenital pulmonary airway malformation (CPAM) type II. e-Figure 53-10 Congenital pulmonary airway malformation type II. Postnatal imaging findings of CPAMs usually correlate with underlying histopathologic features.17 Large cyst type or type I CPAMs typically present with one or several larger air-filled cystic structures with intervening solid, unaerated lung parenchyma. The cysts of type I CPAMs are larger than 2 cm and may be accompanied by several microcysts, whereas small cyst type or type II CPAMs usually manifest as partially air-filled multicystic masses, with individual cysts smaller than 2 cm and with variable degrees of solid-appearing, unaerated lung tissue.10,17 Type 3 CPAMs typically appear as solid lesions with mild contrast enhancement because of microscopic cysts that can be identified only at histologic evaluation. Type IV CPAMs usually present as large cysts arising from the peripheral portion of the lung and can be radiographically indistinguishable from a predominantly cystic type 1 pleuropulmonary blastoma (see Chapter 55) (e-Fig. 53-11). e-Figure 53-11 A cystic pleuropulmonary blastoma in a 6-month-old with increased work of breathing. In their pure forms, CPAM blood supply is from the pulmonary artery and venous drainage is into the pulmonary veins. Although unilobar involvement of CPAM is far more common, multilobar and even bilateral lung involvement may occur.3,27,30 Although any lobe of the lung can be involved, predilection exists for the lower lobe.3 CPAMs that are complicated as a result of superimposed infection may have an imaging appearance similar to pneumonia or a lung abscess (Fig. 53-12). Figure 53-12 An infected congenital pulmonary airway malformation (CPAM) type I lesion in a 15-year-old girl. Treatment and Follow-up: The generalized consensus is that symptomatic CPAMs should be resected, typically by lobectomy, regardless of the patient’s age at presentation.17,22,33 However, considerable controversy exists with regard to the management of prenatally diagnosed, asymptomatic, small CPAM lesions, and no consensus exists on the timing of34 or need for resection.35–39 Although some persons advocate a nonsurgical strategy with imaging follow-up, most medical centers advocate surgical resection before 1 year of age because of the potential risk of associated complications, such as infection, pneumothorax, and the small risk of malignant transformation, particularly in the case of CPAM type I lesions.3,21,22,33,40 Anomalies of the Pulmonary Artery Pulmonary Agenesis, Aplasia, and Hypoplasia Etiology: Pulmonary underdevelopment may be classified into three main types: (1) lung agenesis, consisting of the absence of the lung, bronchus, and pulmonary artery; (2) lung aplasia, that is, the presence of a rudimentary bronchus but the lack of lung tissue and pulmonary artery; and (3) lung hypoplasia, which consists of a hypoplastic bronchial tree and pulmonary artery with a variable amount of lung parenchyma.17,41 The etiology of lung agenesis or aplasia remains uncertain. Genetic, teratogenic, and mechanical factors may play a role.17 Pulmonary agenesis associated with ipsilateral radial ray defects or hemifacial microsomia may be the result of an abnormal development of the first and second arch derivatives or abnormal blood flow at this level inciting the developmental event, given the common association.42 On the other end of the spectrum, no identifiable cause has been found for lung hypoplasia.17 Persons with pulmonary agenesis, aplasia, and hypoplasia either are asymptomatic or present with variable degrees of respiratory distress, depending on the extent of lung underdevelopment. Associated congenital malformations may be seen in 50% to 80% of cases involving the heart, gastrointestinal tract, skeleton, and vascular and genitourinary systems.17,42–44 Imaging: On chest radiographs, affected pediatric patients may or may not present with a small, radiopaque hemithorax, depending on the degree of the abnormality. Ipsilateral displacement of mediastinal structures and elevation of the hemidiaphragm usually are present. The normal contralateral lung shows compensatory hyperinflation and herniation across the anterior midline, which is best seen on the lateral projections as a band of increased retrosternal lucency17 (Fig. 53-13). Left lung agenesis is more common than right lung agenesis. Multidetector CT with multiplanar 2D and 3D imaging capabilities can be used to distinguish among pulmonary agenesis, pulmonary aplasia, and pulmonary hypoplasia by clearly identifying the bronchial stump and/or the rudimentary bronchial tree17,45 (e-Figs. 53-14 and 53-15, Fig. 53-16, and e-Fig. 53-17). Figure 53-13 Pulmonary agenesis. Figure 53-16 Hypoplastic right lung and scimitar syndrome in an 8-year-old. e-Figure 53-14 A 1-day-old with lung aplasia, esophageal atresia, and distal tracheoesophageal fistula. e-Figure 53-15 Ex-34 weeks premature with esophageal atresia, distal tracheoesophageal (TE) fistula, and imperforate anus.
Congenital Lung Anomalies
Spectrum of Congenital Lung Anomalies
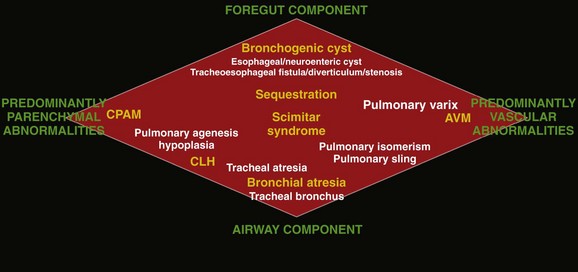
The lesions in yellow denote those most commonly encountered in this entity, whereas the lesions in white represent additional lesions that can be considered part of the spectrum. AVM, Arteriovenous malformation; CPAM, congenital pulmonary airway malformation; CLH, congenital lobar hyperinflation. (Adapted from Newman B. Congenital bronchopulmonary foregut malformations: concepts and controversies. Pediatr Radiol. 2006;36:773-791.)
Predominantly Parenchymal Lesions
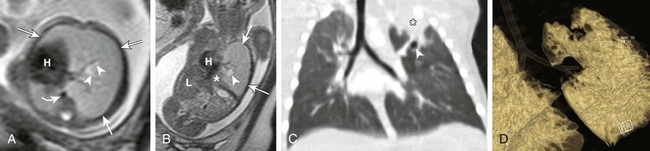
Axial (A) and coronal (B) T2-weighted fetal magnetic resonance images in a 22-week gestational age fetus demonstrate a large homogeneous lesion in the left upper lobe (arrows). Central fluid-filled bronchi are noted (arrowheads). The left lower lobe (asterisk) is compressed inferiorly and displaced medially adjacent to the fetal heart (H). The curved arrow denotes the aorta, which is also displaced to the right. L, Liver. C, A coronal multiplanar reconstruction computed tomography (CT) image shows partial opacification (asterisk) of the left upper lobe, presumably due to mucostasis related to bronchial atresia. In addition, a small air bubble is seen (arrowhead), reflecting a bronchocele. D, A volume-rendered reconstructed CT image could not delineate the left upper lobe bronchus.
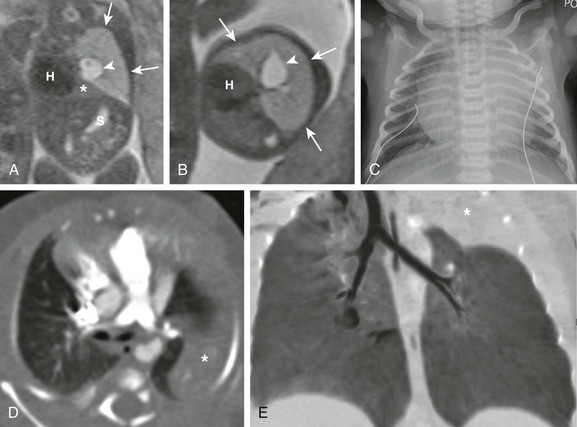
Coronal (A) and axial (B) T2-weighted fetal magnetic resonance images show a large hyperintense, homogeneous lesion in the left upper lobe (arrows) in a 21-week gestational age fetus. The left lower lobe (asterisk) is displaced and compressed inferiorly. The heart (H) is shifted to the right. Central fluid-filled bronchi are noted (arrowheads). S, Stomach. C, A frontal chest radiograph at birth shows complete opacification of the left upper lobe, presumably as a result of retained fetal fluid and mucostasis related to bronchial atresia. The fluid-filled central cystic structure reflects a large mucocele/bronchocele. Axial computed tomography (CT) angiography (D) and minimum intensity projection (E) images performed at day 2 of life show complete collapse of the left upper lobe (asterisk). In addition, the left upper lobe bronchus could not be delineated, a finding suggestive of bronchial atresia. Three-dimensional reconstructions are invaluable in showing the “interrupted connection” to the remainder of the airway.
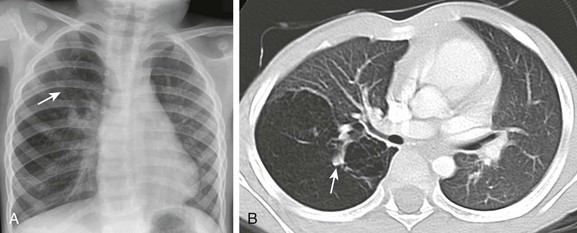
Frontal chest radiograph (A) and axial computed tomography (B) images in a 3-year-old show right upper lobe hyperlucency and branching linear density (arrows) reflecting mucoid impaction in the bronchi distal to the atresia.
Bronchogenic Cysts
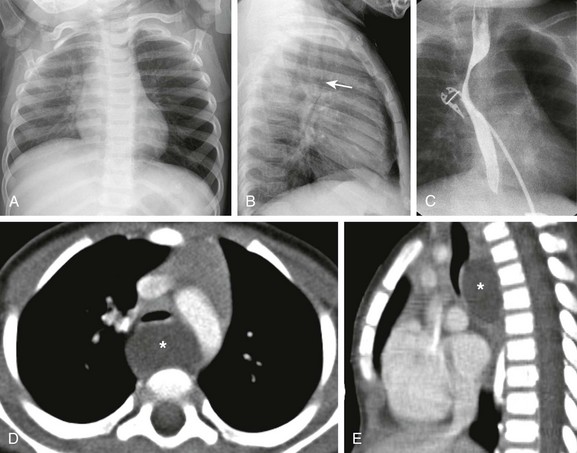
The frontal chest radiograph (A) shows hyperinflation of the left lung. The lateral radiograph (B) reveals the effect of the mass and posterior displacement of the airway (arrow). C, An upper gastrointestinal image shows an esophageal displacement by a soft tissue density mass. Enhanced axial (D) and sagittal (E) computed tomography images show a fluid density lesion (asterisks) posterior to the lower trachea consistent with a bronchogenic cyst.
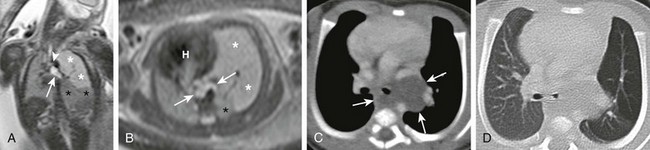
Coronal (A) and axial (B) T2-weighted fetal magnetic resonance images in a 26-week gestational age fetus show a cystic structure in the posterior mediastinum (arrows) apparently compressing the left mainstem bronchus (arrowhead) and resulting in hyperexpansion with increased fluid signal in the left upper lobe (white asterisks). The left lower lobe is compressed and displaced inferiorly (black asterisks). The fetal heart (H) and mediastinum have shifted to the right. Enhanced computed tomography images in mediastinal (C) and lung (D) windows show the fluid-filled cystic structure consistent with a bronchogenic cyst (arrows) centered at the left hilum and extending posteriorly and inferiorly. The cyst compresses the left mainstem bronchus and results in air trapping in the left upper lobe.
Congenital Lobar Hyperinflation
Congenital Pulmonary Airway Malformation
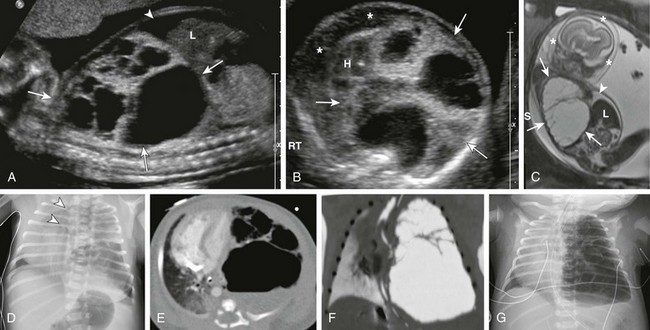
Sagittal ultrasound (A), axial ultrasound (B), and fetal T2-weighted magnetic resonance sagittal (C) images in a 22-week gestational age fetus show a large, heterogeneous, multicystic lesion occupying the vast majority of the left hemithorax (arrows) and resulting in inversion of the left hemidiaphragm. Note the ascites in the abdomen (arrowhead) and the skin thickening and edema (asterisks), which are reflective of hydrops. H, Heart; L, liver. D, A radiograph immediately after delivery shows the complex, partially aerated lesion resulting in significant mediastinal shift. The arrowheads denote the severe tracheal deviation. Axial computed tomography angiography (E) and minimum intensity projection (F) images obtained the following day show the large, heterogenous lesion with multiple macrocysts of varying sizes. Fluid is identified in some of the cysts. G, As fetal fluid is cleared from the lungs, the lesion appears more aerated and further mediastinal shift occurs.
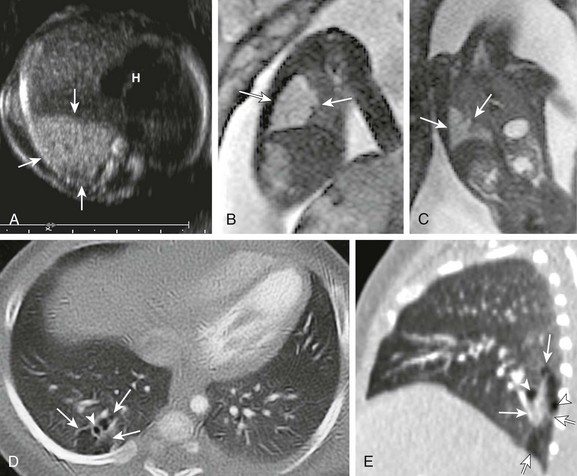
A, A transverse fetal ultrasound image through the chest in a 20-week gestational age fetus shows an echogenic right lower lobe lung lesion with tiny cysts only seen on high-resolution images (image not shown) consistent with a CPAM type II lesion (arrows). The heart (H) and mediastinum are only mildly shifted without cardiac compression. Sagittal (B) and coronal (C) fetal T2-weighted magnetic resonance images show a mildly heterogenous lesion (arrows) with slightly nodular contours but no visible cysts. Postnatal axial and sagittal computed tomography images (D and E) at 1 month of age show an ill-defined, relatively smaller lesion (arrows) in the right lower lobe with tiny, subcentimeter internal cysts (arrowheads). This lesion also demonstrated bronchial atresia components on histologic examination.
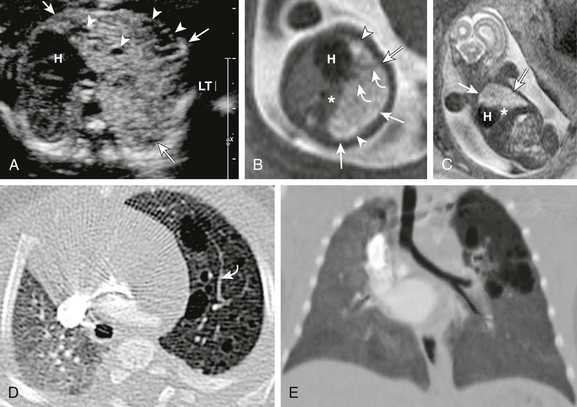
A, A transverse fetal ultrasound image through the chest in a 19-week gestational age fetus shows an echogenic left upper lobe lesion with several internal cysts (arrowheads) and findings consistent with a CPAM type 2 lesion (arrows). The heart (H) and mediastinum are moderately shifted to the right without cardiac compression. LT, left. Axial (B) and coronal (C) fetal T2-weighted magnetic resonance (MR) images show a heterogeneous lesion (arrows) with internal cysts (arrowheads); some of them even appear as “macrocysts.” Normal-appearing lung tissue is noted as being compressed posteriorly and inferiorly (asterisks). However, on postnatal axial (D) and minimum intensity projection (E) computed tomography (CT) images at 3 months of age, the lesion appears relatively smaller and exerts less mass effect and no mediastinal shift. All the cysts are aerated and measure less than 2.0 cm. Note that some slightly prominent, tortuous internal pulmonary vessels are seen within the lesion in both the fetal MR image (B) and the postnatal CT image (D) (curved arrows).
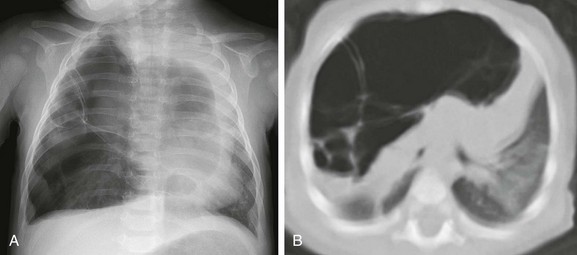
A, A frontal chest radiograph demonstrates a large radiolucent lesion with fine septations and multiple cysts within the right lung resulting in a mediastinal shift. B, An axial computed tomography image reveals multiple cysts of varying sizes with effect of the mass demonstrated on the mediastinum and contralateral left lung.
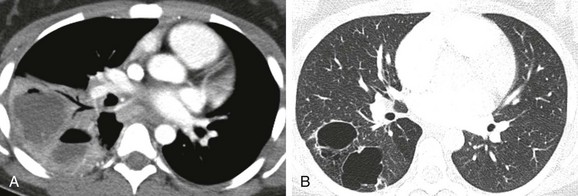
A, An initial computed tomography (CT) enhanced image shows consolidation, with areas of low attenuation representing superimposed infection of the cystic components. B, A follow-up CT image obtained 7 months later shows resolution of the infection and visualization of CPAM cystic components.
Predominantly Vascular Lesions
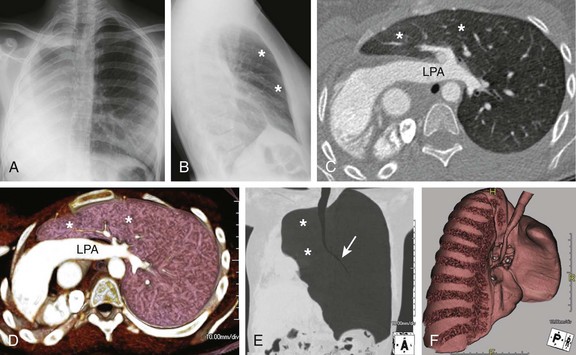
Frontal (A) and lateral (B) radiographs in a 15-year-old with worsening asthma demonstrate marked hyperinflation of the left lung that extends across the midline anteriorly and herniates toward the right, as evidenced by a band of retrosternal lucency on the lateral projection (asterisks). Associated dextroposition of the heart into the right hemithorax is seen. Enhanced axial computed tomography (C), axial volume-rendered (D), minimum intensity projection (E) and three-dimensional volume-rendered (F) images of the central airway and lungs demonstrate complete agenesis of the right bronchus and lung. Associated dextroposition of the heart and compensatory hyperexpansion is present, particularly of the left upper lobe (asterisks), which herniates into the right hemithorax. A normal left mainstem bronchus (arrow) is seen. LPA, Left pulmonary artery.
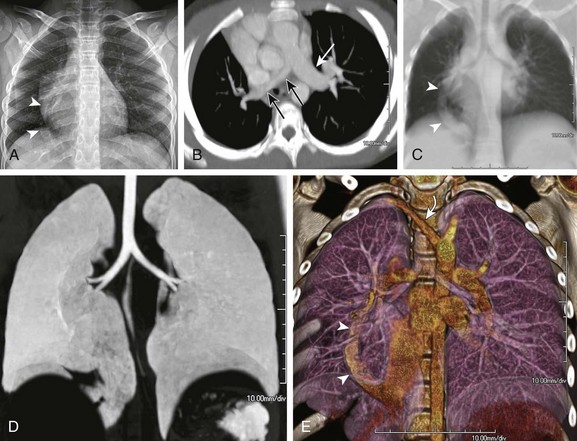
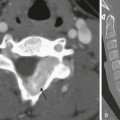
A, A frontal chest radiograph shows asymmetric lung volumes with dextroposition of the heart and a vertically oriented curvilinear opacity (arrowheads) projecting over the right lower hemithorax. An axial maximum intensity projection computed tomography image (B) confirms mild dextroposition of the heart and hypoplastic right pulmonary artery (PA) (black arrows) relative to the normal left pulmonary artery (white arrow). Coronal thick multiplanar reconstruction (C), inverted minimum intensity projection (D), and three-dimensional volume rendered (E) images show hypoplasia of the right lung with absence of the right upper lobe bronchus and partial anomalous pulmonary venous return of a vast portion of the right lung via a scimitar vein (arrowheads) into the inferior vena cava. Incidentally noted is a right aberrant subclavian artery (curved arrow).
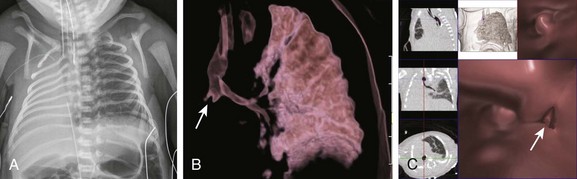
A, A frontal chest radiograph shows complete opacification with volume loss in the right hemithorax. The airway and mediastinum are shifted to the right with associated dextroposition of the heart. A nasogastric tube terminates in the upper esophageal pouch consistent with esophageal atresia. The anterior view of a volume-rendered image of the central airway and lungs (B) and virtual bronchoscopic (C) images demonstrate a rudimentary right main stem bronchus (straight arrows) consistent with lung aplasia. No right pulmonary artery is identified (image not shown).
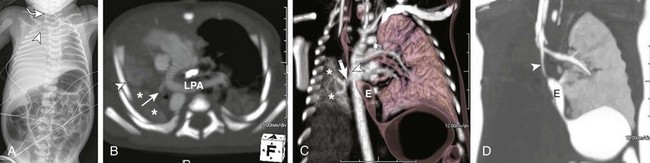
A, A frontal radiograph of the chest and abdomen shows complete opacification with volume loss in the right hemithorax. The endotracheal tube (arrowhead) and mediastinum are shifted to the right with associated dextroposition of the heart. A nasogastric tube (curved arrowRelated posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree