Connective tissue diseases (CTDs) may be associated with a variety of lung abnormalities. Most patients with CTD have extra-pulmonary manifestations but in some cases, lung abnormalities are seen in isolation or as the first manifestation of disease.
This chapter provides an overview of the approach to diagnosis of lung disease in these patients, followed by a discussion of the most common lung manifestations of specific CTDs.
A GENERAL APPROACH TO DIAGNOSIS
CTDs may be manifested by a number of pulmonary abnormalities that reflect the various ways the lung reacts to injury (Table 10.1). The interstitial pneumonias (IPs) are common in patients with CTD, including usual interstitial pneumonia (UIP), nonspecific interstitial pneumonia (NSIP), lymphoid interstitial pneumonia (LIP) and follicular bronchiolitis, organizing pneumonia (OP), and diffuse alveolar damage (DAD). While patients tend to present with a single pattern, an overlap of more than one pattern is sometimes seen. The IPs are described in detail in the preceding chapter.
In addition, patients with CTD may show intrathoracic abnormalities not related to an IP. These may be specific to the individual CTD. These include pulmonary edema, vasculitis, pulmonary hypertension, pulmonary hemorrhage, pleural or pericardial effusion, lung nodules, bronchiectasis, constrictive bronchiolitis, and esophageal dilatation.
The initial presentation of diffuse lung disease may precede the diagnosis of CTD. HRCT plays a major role in suggesting the presence of a pattern of lung disease (e.g., NSIP) consistent with CTD, and excluding alternative diagnoses. In some patients, HRCT findings precipitate a workup for CTD.
HRCT may also be used to categorize the pattern of lung disease present in patients with known CTD. This is important for treatment and determining prognosis. Serologies are also commonly obtained to suggest etiology in a patient with undiagnosed diffuse lung disease or for further classification of the CTD present. The most useful serologies are those that are relatively specific for a particular CTD, although no serology is diagnostic of a single disease. For instance, the serology Scl-70 is relatively specific for a diagnosis of scleroderma.
Table 10.1 Abnormalities that may be seen in patients with connective tissue disease
Usual interstitial pneumonia
Nonspecific interstitial pneumonia
Lymphoid interstitial pneumonia
Follicular bronchiolitis
Organizing pneumonia
Diffuse alveolar damage
Constrictive bronchiolitis
Pulmonary edema
Pulmonary hemorrhage
Pulmonary hypertension
Serositis (pleural or pericardial effusion)
Miscellaneous (nodules, bronchiectasis, esophageal dilatation)
Any CTD may be present with any of the HRCT patterns listed above; however, some general trends exist. When diffuse lung disease is present in a patient whose diagnosis of CTD is established, it is usually assumed to be a manifestation of that systemic disorder unless there is convincing evidence to the contrary. For this reason, patients with CTD uncommonly undergo biopsy and HRCT is often the primary diagnostic examination that determines the pattern of lung disease.
PATTERNS OF DIFFUSE LUNG DISEASE
As HRCT is the primary method of characterizing diffuse lung disease in patients with CTD, an in-depth knowledge of their manifestations and differentiating features is important.
UIP and NSIP
UIP and NSIP are the most common causes of diffuse lung disease and fibrosis in patients with CTD. Typical findings (Table 10.2) include honeycombing, traction bronchiectasis, and irregular reticulation. Ground glass opacity is less common, but may be seen in NSIP, particularly the cellular subtype. Fibrosis is only rarely seen with LIP and follicular bronchiolitis, OP, and constrictive bronchiolitis.

In general, NSIP is the most common pattern seen in patients with CTD. It is most typical of scleroderma, polymyositis, dermatomyositis, and mixed CTD. UIP is seen most frequently in patients with rheumatoid lung disease.
UIP and NSIP may be difficult to differentiate radiographically, unless ground glass opacity or subpleural sparing is present. In particular, the fibrotic subtype of NSIP can closely resemble UIP, although the clinical significance of this distinction is uncertain. In UIP, honeycombing is a significant finding (Fig. 10.1), whereas in NSIP, it is either absent or minimal in extent (Fig. 10.2). The greater the severity of fibrosis in the absence of honeycombing, the more likely NSIP becomes. Subpleural sparing strongly suggests NSIP over UIP (Fig. 10.3).
LIP and Follicular Bronchiolitis
LIP and follicular bronchiolitis are thought to represent different points on a spectrum of the lymphocytic lung infiltration; thus, their findings may overlap. The typical findings of LIP (Table 10.3) include patchy ground glass opacity, centrilobular or perilymphatic nodules, and, to a lesser extent, consolidation. Lung cysts may be seen in association with other findings (Fig. 10.4) or as an isolated abnormality. Cysts are typically thin walled and limited in number and vessels may be seen in association with their walls.
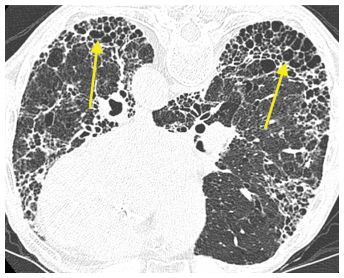
Figure 10.1
Usual interstitial pneumonia in rheumatoid arthritis. Prone HRCT shows peripheral and basilar fibrosis with extensive honeycombing (arrows) typical of usual interstitial pneumonia. While this may be seen with any connective tissue disease, it is most common with rheumatoid lung disease.
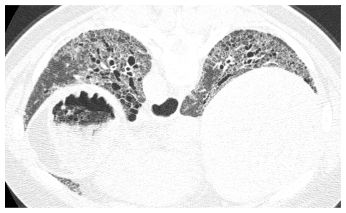
Figure 10.2
Nonspecific interstitial pneumonia (NSIP) in scleroderma. Basilar predominant fibrosis is present with irregular reticulation and traction bronchiectasis. No honeycombing is seen. In the setting of known connective tissue disease, this is compatible with fibrotic NSIP, and a biopsy is not usually required for diagnosis.
Follicular bronchiolitis represents localized lymphoid infiltration of bronchioles. It commonly presents with centrilobular nodules of ground glass opacity (Fig. 10.5). Air trapping and mosaic perfusion may also be present. Lung cysts are thought to reflect the presence of air trapping associated with follicular bronchiolitis.
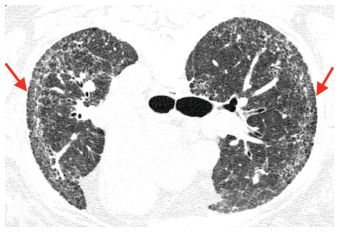
Figure 10.3
Nonspecific interstitial pneumonia (NSIP) with subpleural sparing. Prone HRCT shows irregular reticulation and traction bronchiolectasis in the peripheral lung, without significant honeycombing. Sparing of the immediate subpleural lung (arrows) is highly suggestive of NSIP. While the irregular reticulation likely represents irreversible disease (fibrotic NSIP), some of these abnormalities may resolve with treatment.
Table 10.3 HRCT findings of lymphoid interstitial pneumonia and follicular bronchiolitis
Centrilobular nodules of ground glass opacity Perilymphatic nodules
Patchy, bilateral ground glass opacity Patchy, bilateral consolidation
Lung cysts (thin walled, limited in number)
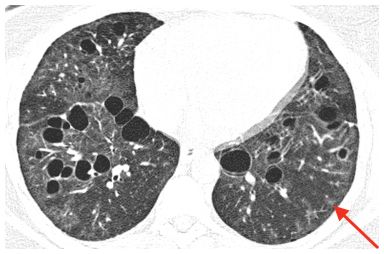
Figure 10.4
Lymphoid interstitial pneumonia. Cysts are seen in association with ground glass opacity in a patient with polymyositis. Small areas of ground glass opacity appear to have a centrilobular distribution (arrow).
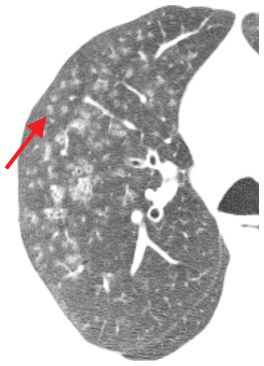
Figure 10.5
Follicular bronchiolitis. Centrilobular nodules of ground glass opacity are noted, associated with mild bronchial dilatation. Note that the nodules are located at a distance from the pleural surfaces (arrow) and are evenly spaced from one another. This is typical of a centrilobular distribution.
Table 10.4 HRCT findings of organizing pneumonia
Consolidation, focal, irregular borders, often nodular or mass-like
Ground glass opacity (uncommon)
Centrilobular nodules (rare)
Peribronchovascular/subpleural predominant
Atoll or reversed halo sign
Among the CTDs, LIP and follicular bronchiolitis are most commonly seen in patients with Sjögren’s disease. Isolated cysts in the setting of Sjögren’s disease are highly suggestive of LIP. LIP and follicular bronchiolitis may also be seen in combination with other patterns of lung disease. For instance, patients with NSIP may have centrilobular nodules in the peripheral lung representing follicular bronchiolitis.
In the absence of signs of fibrosis, ground glass opacity is not commonly seen with the other patterns of chronic lung disease in CTD, with the exception of NSIP. NSIP is typically peripheral and basilar in distribution, whereas LIP and follicular bronchiolitis usually show involvement of the central lung regions. Chronic ground glass and centrilobular nodules may be seen with diseases unrelated to CTD such as hypersensitivity pneumonitis and smoking-related lung disease.
Organizing Pneumonia
OP typically presents with focal areas of consolidation with irregular borders that often appear nodular or mass-like (Table 10.4). The distribution of consolidation is typically peribronchovascular and/or subpleural (Fig. 10.6A, B). Ground glass opacity is less common and typically seen in immunosuppressed patients. Centrilobular nodules are a rare finding. Architectural distortion and mild bronchial dilatation may be seen associated with these opacities. After treatment, OP may result in mild scarring; however, extensive fibrosis resembling NSIP or UIP is rare.
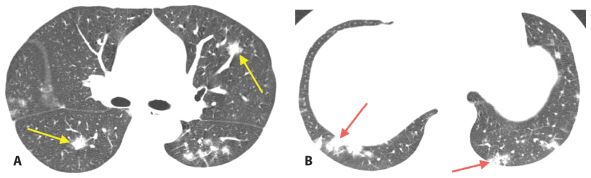
Figure 10.6
Organizing pneumonia. Patchy peribronchovascular (A, arrows) and subpleural (B, arrows) consolidation is seen in a patient with organizing pneumonia due to systemic lupus erythematosus–associated lung disease.
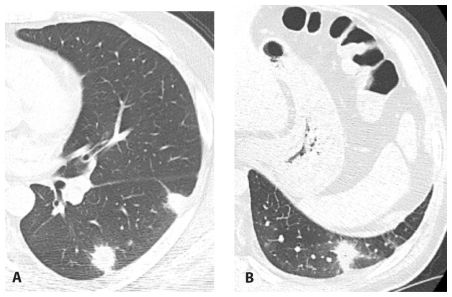
Figure 10.7
Fungal infection resembling organizing pneumonia in a patient with connective tissue disease. HRCT images through the mid-lung (A) and lung base (B) show large ill-defined nodules from fungal infection that have an appearance similar to organizing pneumonia.
Related posts:
 HRCT Indications, Technique, Radiation Dose, and Normal Anatomy
HRCT Indications, Technique, Radiation Dose, and Normal Anatomy
 Pulmonary Edema, Diffuse Alveolar Damage, the Acute Respiratory Distress Syndrome, and Pulmonary Hemorrhage
Pulmonary Edema, Diffuse Alveolar Damage, the Acute Respiratory Distress Syndrome, and Pulmonary Hemorrhage
 Hypersensitivity Pneumonitis and Eosinophilic Lung Disease
Hypersensitivity Pneumonitis and Eosinophilic Lung Disease
 Complications of Medical Treatment:Drug-Induced Lung Disease and Radiation
Complications of Medical Treatment:Drug-Induced Lung Disease and Radiation
 Reticular Opacities
Reticular Opacities
 Nodular Lung Disease
Nodular Lung Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


