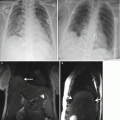
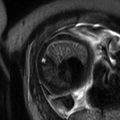
Fig. 5.1
Giant cervical lymphangioma. Coronal (a), sagittal (b), and axial (c) HASTE MR images demonstrate a large anterior multicystic neck mass (arrow), with no compression to the airways in a fetus at 27 weeks’ gestation. Orotracheal intubation (d) under direct laryngoscopy performed during EXIT, i.e., by preserving uteroplacental blood flow. Note the partial delivery of the fetus through the hysterotomy and the untouched umbilical cord (asterisk). The fetus is monitored with a pulse oximeter attached to his left hand
5.2.3 Cervical and Mediastinal Teratoma
The head and neck is the second commonest site of teratoma in the fetus [8]. These masses typically consist of both cystic and solid components and originate from the palate, nasopharynx, or thyrocervical area. The finding of calcifications is nearly diagnostic of teratoma, although these are present only in approximately 50 % of cases and might not be conspicuous on USS or MRI [8]. A common and important associated finding seen with cervical and oral teratomas is the polyhydramnios, resulting from a direct mass effect, because swallowing can be compromised by occlusion of the mouth or compression of the esophagus [8].
The management of fetal giant cervical teratoma includes a spectrum of options. For the rare fetus who develops hydrops fetalis, fetal resection may be indicated [10]. In patients with airway obstruction, EXIT procedure provides the luxury of time to obtain airway control either by intubation, tracheostomy, or, if necessary, tumor resection on placental support [11].
Fetal mediastinal teratomas are rare tumors that, if massive in size, may cause hydrops leading to fetal demise or fetal esophageal and airway compression causing late-gestation polyhydramnios and preterm labor. In some instances, the occurrence of hydrops fetalis has been managed successfully by in utero aspiration of the tumor cyst fluid and postnatal tumor resection [12] or by EXIT procedure for establishment of an airway and tumor resection on uteroplacental support [13].
Fetal MRI has proven to be a useful complementary imaging tool to characterize cervical and mediastinal teratoma, helping to direct optimal prenatal and perinatal management of these lesions (Fig. 5.2).
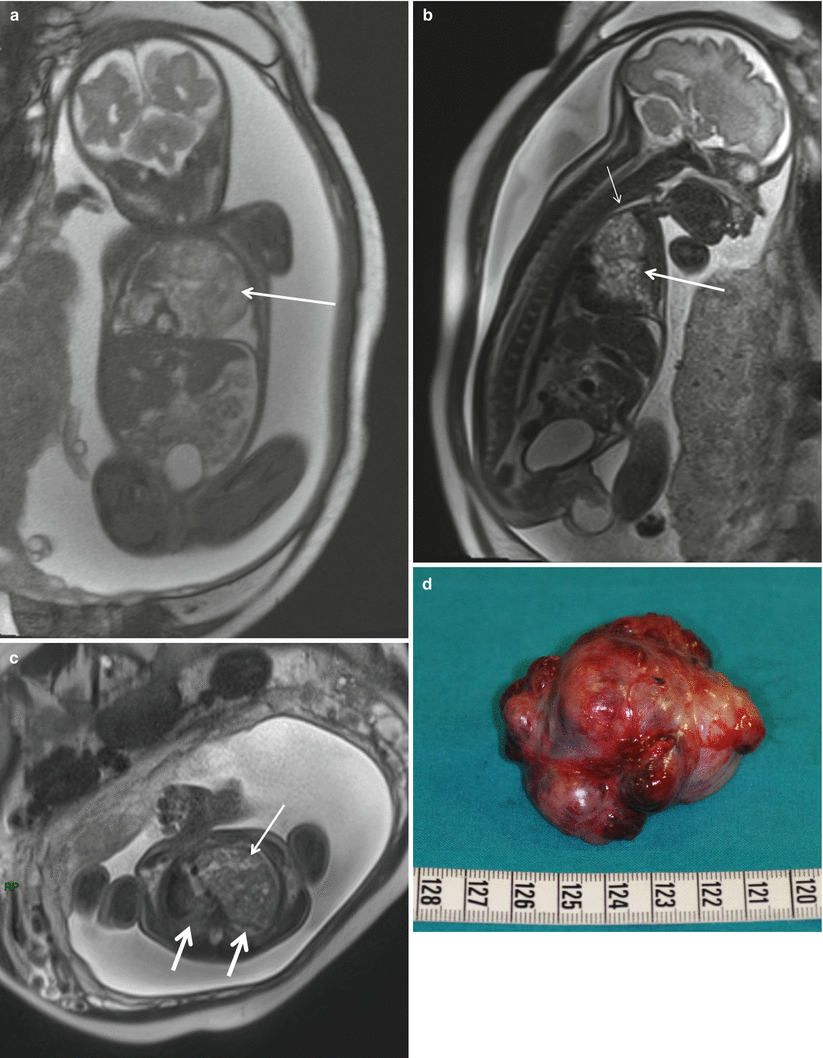
Fig. 5.2
Giant mediastinal teratoma. Coronal True Fisp (a), sagittal (b), and axial (c) T2-weighted HASTE MR sequences demonstrate a large heterogeneous mediastinal mass (arrow) that deviates the heart to the right in a fetus at 27 weeks’ gestation. Note the small lungs of relatively low signal intensity (thick arrows in c). Note pleural effusion, ascites, and generalized subcutaneous edema. The airway below the mass is dilated (short arrow in b). Macroscopic appearance (d) of the surgical resection specimen of the tumor
5.3 Congenital Diaphragmatic Hernia (CDH)
Congenital diaphragmatic hernia (CDH) is a common surgically correctable defect, occurring in 1:3000 pregnancies. Despite great strides in its management made in recent years, CDH continues to be associated with significant morbidity and mortality.
This disorder is characterized by a defect in the diaphragm allowing herniation of the abdominal viscera into the thorax that compromise normal lung development. The current mainstay of treatment of CDH involves stabilization and respiratory support at birth, followed by closure of the diaphragmatic defect, which returns the abdominal organs to the abdominal cavity and makes room in the chest for the hypoplastic lung to grow. Despite some recent series from specialized centers reporting a survival rate of close to 90 %, there is a significant divergence in survival data for CDH due to potential case selection bias, primarily because as many as 35 % of live-born infants with CDH do not survive to transport resulting in a “hidden mortality.” Hidden mortality is referred to patients who die before surgery, either during gestation or shortly after birth, and thus are not reported by individual institutions. Specific morbidities in survivors include neurodevelopmental, nutritional, sensorineural hearing, and pulmonary function deficiencies, all of which are most likely attributable to the severity of lung hypoplasia and pulmonary hypertension that accompany CDH [1, 2]. Marked hypoplasia and persistent pulmonary hypertension, in fact, complicate postnatal management in severe cases and are the justification for considering antenatal intervention in selected cases.
Now that two-thirds of CDH can be accurately diagnosed by mid-gestation, multiple attempts have been made to find accurate prenatal parameters that predict neonatal outcomes with much of the focus on measurements of lung size and vasculature, as well as on intrathoracic liver herniation.
The most commonly used parameter used to predict neonatal outcomes is the lung area to head circumference ratio (LHR), which is a sonographically obtained measurement reflecting the relative lung size contralateral to the diaphragmatic defect. Unfortunately, this parameter is highly operator dependent, can be measured in various formats, and has gestational limitations that make it only reliable when measured between 22 and 28 weeks’ gestation in the presence of liver herniation [14]. These limits have been obviated by using the observed to expected LHR (o/e LHR). However, its sensitivity in predicting neonatal survival in left-sided CDH is still only 46 % with 10 % false-positive rate, likely a limitation of the two-dimensional measurement [15]. Even with advances in three-dimensional USS, the contralateral lung volume is underestimated by 25 % with the ipsilateral lung not even visualized for measurement in nearly 45 % of evaluation.
Fetal magnetic resonance imaging (MRI) is an alternative imaging study with improved accuracy in fetal lung volume measurement. This modality provides improved soft-tissue contrast and is not affected by maternal body habitus or fetal position. Some studies have demonstrated total lung volume (TLV), observed to expected TLV (o/e TLV), and the percentage of predicted lung volume (PPLV) to be predictive of neonatal outcomes while alleviating some of the shortcomings of USS [16–21]. Busing et al. have demonstrated that TLV alone has acceptable prognostic ability to predict neonatal survival and need for extracorporeal membrane oxygenation (ECMO), comparable with o/e TLV [16]. PPLV, which evaluates TLV in regard to the expected volume of the individual fetus, has also been shown to predict ECMO use, length of hospital stay, and neonatal survival [22]. Generally, fetal lung volume increases considerably in late gestation [23]. Bargy et al. analyzed postmortem lungs from CDH fetuses and found that pulmonary hypoplasia became progressively worse with advancing gestation, especially beyond 30 weeks’ gestation [24].
Coleman et al. have recently hypothesized that fetal lung growth rate is related to neonatal outcome. In their longitudinal study of isolated left CDH fetuses, a significant difference in lung growth (rate of change in TLV) was found between survivors and non-survivors [25]. The authors found that the severity of pulmonary hypoplasia is dynamic and can worsen in the third trimester. Therefore, they concluded that all prenatal MRI-derived lung volumes are more predictive of outcome closer to term, facilitating prenatal counseling and focusing perinatal management.
MRI can also demonstrate the presence of intrathoracic liver herniation and quantify the degree of that herniation (Fig. 5.3). However, there is no clear consensus on the landmarks to define/quantify liver herniation at present. Although current published data suggest but are not conclusive whether the liver is truly an independent predictor of outcome, next to lung size measurements, at present, both variables should be considered when counseling the patient.
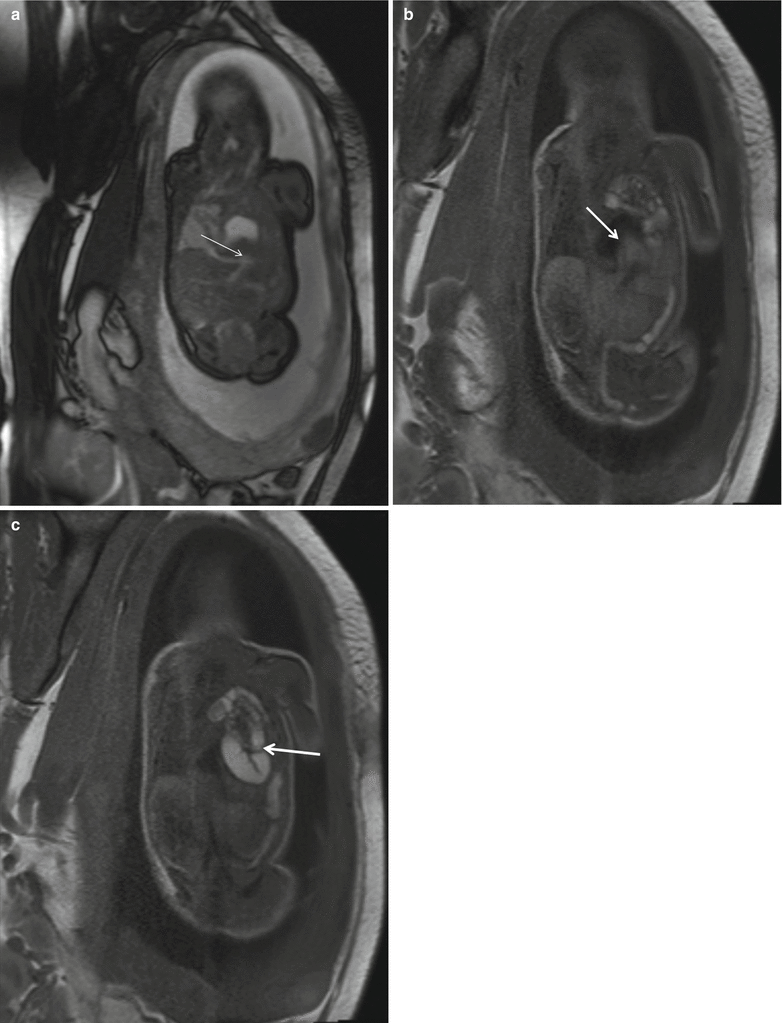
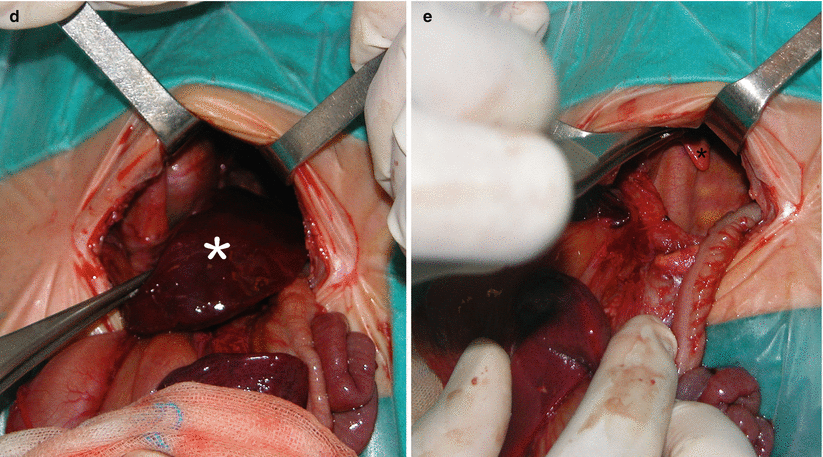
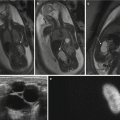
Fig. 5.3
Prenatal diagnosis of left-sided CDH with intrathoracic liver herniation. Coronal True Fisp (a) and T1-weighted (b, c) MR images show upward herniation of part of the liver into the chest across the diaphragmatic defect of a left-sided CDH defect in a fetus at 24 weeks’ gestation. Note the dark T2 signal of the liver compared with the higher T2 signal of the lung. Note also the hyperintense signal of the herniated colon (arrow in c). Intraoperative view (d) of the “liver up” (asterisk) before its reduction into the abdominal cavity. Intraoperative view (e) of the left-sided CDH following complete reduction of herniated viscera. Note the markedly hypoplastic lung (asterisk) and the wide size of the defect that required patch repair
Novel methods, in particular those evaluating the pulmonary circulation – either by measurements at baseline or following maternal hyperoxygenation – as well as MRI diffusion imaging might help in the prediction of the second cause of death in CDH, which is pulmonary hypertension. A great advantage of USS is the possibility to visualize and measure blood flow without the need for contrast agents. Ruano et al. investigated the predictive value of 3D power Doppler of the entire lung vasculature in terms of survival as well as the occurrence of pulmonary hypertension [26]. Thereafter, other authors found a strong correlation between the reduction in lung tissue perfusion, increased intrapulmonary artery impedance, and lung growth, as evaluated by o/e LHR [27].
Some authors have recently proposed the use of the diffusion-weighted imaging (DWI) for the assessment of fetal lung. DWI is an MRI modality that maps the microstructural characteristics of water diffusion, i.e., the random thermal (Brownian) motion of water molecules in capillaries and extravascular space.
The principal claim is that DWI–MRI might provide functional information of the fetal lung, such as increased interstitial tissue pressure or impaired capillarization. Preliminary work on the use of DWI–MRI as a tool to differentiate between normal and pathological lung development has shown a significant relationship between DWI–MRI parameters and gestational age in the normal fetus but failed to validate DWI–MRI as a tool to predict postnatal outcome in fetuses with CDH [28].
Finally, both USS and MRI can be used to plan the need and the results of fetal interventions for CDH. The first successful fetal surgery for severe CDH was performed at the University of California in San Francisco. Fetal surgery for CDH was originally proposed to involve an “open” approach to repair the diaphragmatic defect in mid-gestation. Indeed, today, there are a handful of survivors of open fetal CDH repair [29].
Thereafter, animal studies, mainly using the fetal lamb model of CDH, showed that temporary occlusion of the fetal trachea prevents the egress of lung fluid, thus promoting lung growth. The development and implementation of minimally invasive surgical techniques advanced at a rapid pace. Simultaneously, irritation of the uterus and the threat of preterm labor were identified as major hurdles in fetal interventions. Initially, a surgical clip was placed on the trachea through a neck incision. Although this technique effectively occluded the trachea, scarring and the development of tracheal stenosis were identified as significant adverse effects. Ultimately, fetal bronchoscopy with tracheal balloon occlusion emerged as an appropriate solution. Further refinements of this technique lead to a novel strategy – percutaneous fetoscopic endoluminal tracheal occlusion (FETO). Small endoscopes and video equipment are used to place a detachable balloon via fetal bronchoscopy around 28 weeks’ gestation with removal of the balloon at 34 weeks’ gestation. The advantage of this treatment is the lack of maternal hysterotomy and its associated morbidity.
After the FETO intervention, both USS and MRI can be used to confirm the adequate position of the balloon and to evaluate lung growth rate [30].
Finally, combination of lung volume parameters and the microstructural fetal lung evaluation using DWI may provide more information on lung growth and maturation in these cases to determine objective time frames for balloon release [28].
The complications after FETO include difficulty in balloon retrieval and postnatal tracheomegaly. However, fetal tracheal occlusion for CDH remains an investigational therapy for which the long-term benefits have yet to be proven.
5.4 Congenital Pulmonary Airway (Cystic Adenomatoid) Malformation
Congenital pulmonary airway malformation (CPAM), previously known as congenital cystic adenomatoid malformation (CCAM), is a rare developmental anomaly of the lower respiratory tract, occurring approximately in 1:30,000 pregnancies. Despite its rarity, CPAM is still the most common congenital lung lesion.
Although the underlying cause for CPAM is unknown, they are thought to result from an early abnormal development of the airway from intrauterine airway obstruction [31], supported by histologic and pathologic changes of exuberant primary bronchiolar overgrowth in communication with an abnormal bronchial tree lacking cartilage.
In CPAM, usually an entire lobe of lung is replaced by a nonworking cystic piece of abnormal lung tissue. This abnormal tissue will never function as normal lung tissue.
Basing on clinical and pathologic features, CPAMs can be classified into five types (revised Stocker classification) that take into account also the size of the lesions:
Type 0: Severe acinar dysgenesis affecting all lung lobes, uniformly fatal.
Type 1: Solitary or multiple macrocysts (>2 cm); this type is of bronchial or bronchiolar origin.
Type 2: Single or multiple cysts of bronchiolar origin, measuring between 0.5 and 2 cm.
Type 3: Multiple microcysts measuring 0.5 cm; they are predominately solid. This type is the only adenomatoid type and has a bronchiolar–alveolar duct origin.
Type 4: Large air-filled cysts, with a distal acinar origin. Notably, they are indistinguishable by imaging from type 1 pleuropulmonary blastoma, appearing as a large cystic lesion.
Large-cyst subtypes account for about 70 % of CPAMs (Fig. 5.4). In most of the cases, the outcome of a fetus with CPAM is very good. However, in rare cases, the cystic mass grows so large as to limit the growth of the surrounding lung and cause pressure against the heart. In these situations, the CPAM can be life-threatening for the fetus.
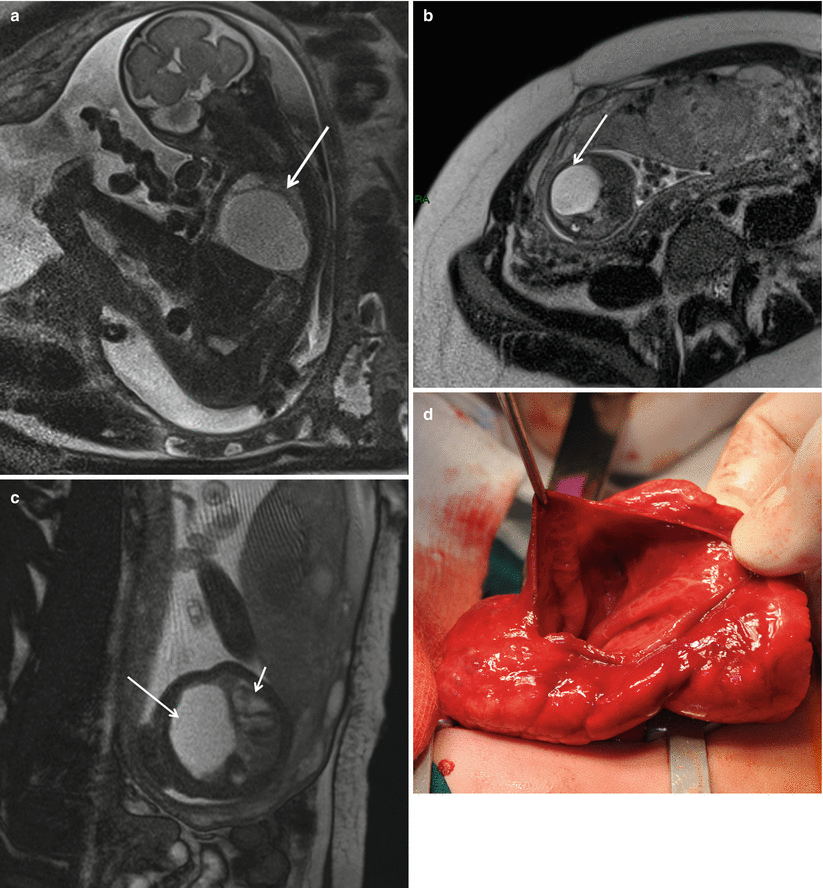
Fig. 5.4
Congenital pulmonary airway (cystic adenomatoid) malformation. HASTE oblique sagittal (a), axial HASTE (b), and True Fisp (c) MR images obtained at 28 weeks’ gestation showing a large high signal mass in the right chest (arrows). The lesion causes mediastinal shift of the heart to the left (short arrow in c). Intraoperative view (d) of a large-cyst subtype CPAM affecting the right lower lobe, requiring lobectomy few days after birth
The prenatal diagnosis of lung pathologies has considerably increased in recent years due to the generalization and technical improvements of USS screening during pregnancy. Prenatal USS may assess the overall mass size and associated compressive effects on the adjacent normal lung and mediastinal structures. Macrocystic CPAMs (types 1 and 2) appear on prenatal USS as echogenic masses with variable-sized cysts, while type 3 CPAMs appear as a homogenous echogenic mass on USS, being indistinguishable from other solid congenital lung anomalies.
Although USS enables a first recognition of the lesions and detection of adverse prognostic factors, their complete characterization in utero may be difficult or inconclusive, mainly because of technical problems, maternal habitus, or fetal position. Complementary fetal MRI has been increasingly performed over the last few years [32–35] as an adjunct to USS in the imaging of fetuses with congenital lung malformations as early as 18 weeks’ gestation [36, 37]. Compared to USS, fetal MRI allows for multiplanar imaging and may be superior at characterization of the boundaries of the malformed lung and how it relates to the normal lung lobar anatomy and surrounding thoracic structures [38–40].
The prenatal natural history of CPAM varies: in many cases, the lesions actually regress, and some disappear completely. Most of the remaining lesions will have a stable prenatal course and can be treated after birth. However, a minority of cases may develop worrisome signs such as mediastinal shift, polyhydramnios, and hydrops fetalis. These events are secondary to the mass effect exerted by the enlarging fetal CPAM. Hydrops, in itself, is harbinger of impending fetal death and is characterized by edema in two or more fetal compartments, e.g., subcutaneous collection, pleural effusion, pericardial effusion, ascites. Therefore, fetuses with congenital lung lesions resulting in a significant intrathoracic mass effect and risk for pulmonary hypoplasia and/or hydrops without other anatomical or chromosomal abnormalities may be candidates for fetal intervention. The mode of intervention is dependent on the type of lesion as well as the gestational age. Open fetal surgery was, and still is, being done for hydropic fetuses, especially for microcystic CPAM lesions. Following fetal lobectomy, survival rates of 60 % have been reported with resolution of hydrops, in utero lung growth, and normal postnatal development [41]. More recently, early delivery with controlled resection of large fetal lung lesions using an EXIT strategy has been performed in hydropic fetuses of greater than 32 weeks’ gestation [42].
However, in utero thoracoamniotic (TA) shunt is the fetal intervention most frequently advocated for CPAM detected before 32 weeks’ gestation and carrying a dismal prognosis [43, 44].
The largest experience with this fetal procedure has been recently published by the Children’s Hospital of Philadelphia (CHOP) group, who reported 97 shunts placed in 75 fetuses [45]. Average gestational age at shunt placement was 25 weeks and survival was 68 %, which depended on GA at birth, reduction in mass size, and hydrops resolution. Surviving infants had prolonged intensive care needs and often required either surgical resection or tube thoracostomy in the perinatal period. Despite the survival benefit, shunt placement is not without the risk of complications including shunt failure by occlusion or migration. Another known fetal complication of shunt placement is the production of chest wall deformities when shunts are placed for large macrocystic CCAMs at 18–20 weeks’ gestation [46]. Finally, an initial experience with the use of steroid therapy given during the second trimester of pregnancy has been successfully reported in fetuses with hydrops fetalis and predominantly microcystic CPAM [47, 48].
Fetuses with CCAM but without hydrops have a good chance for survival with maternal transport and planned delivery for immediate neonatal care. Postnatal treatment options include either a lobectomy or segmentectomy for symptomatic patients. Treatment for asymptomatic patients is still controversial, as some authors suggest an elective resection, given the associated risks of hemorrhage, recurrent infection, and potential risk for malignancy.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


