FIGURE 19.1-1: Relationship of the intermediate mesoderm of the pronephric, mesonephric, and metanephric systems. (From Sadler TW. The urogenital system. In: Sadler TW, ed. Langman’s Medical Embryology. 9th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:321–362.)
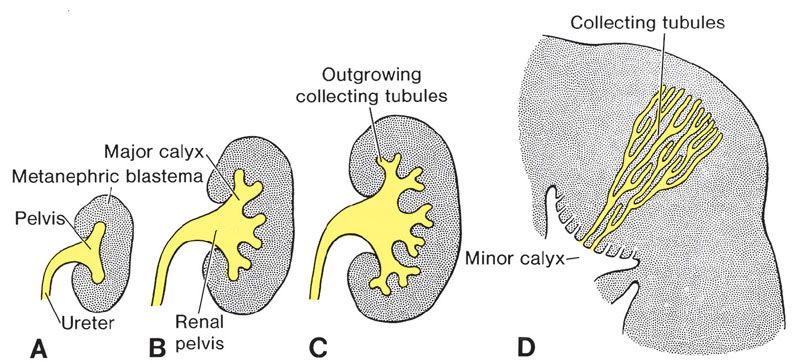
FIGURE 19.1-2: Development of the renal pelvis calyces and collecting tubules of the metanephros at 6 weeks (A), at the end of 6 weeks (B), at 7 weeks (C), and in the newborn (D) . (From Sadler TW. The urogenital system. In: Sadler TW, ed. Langman’s Medical Embryology. 9th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:321–362.)
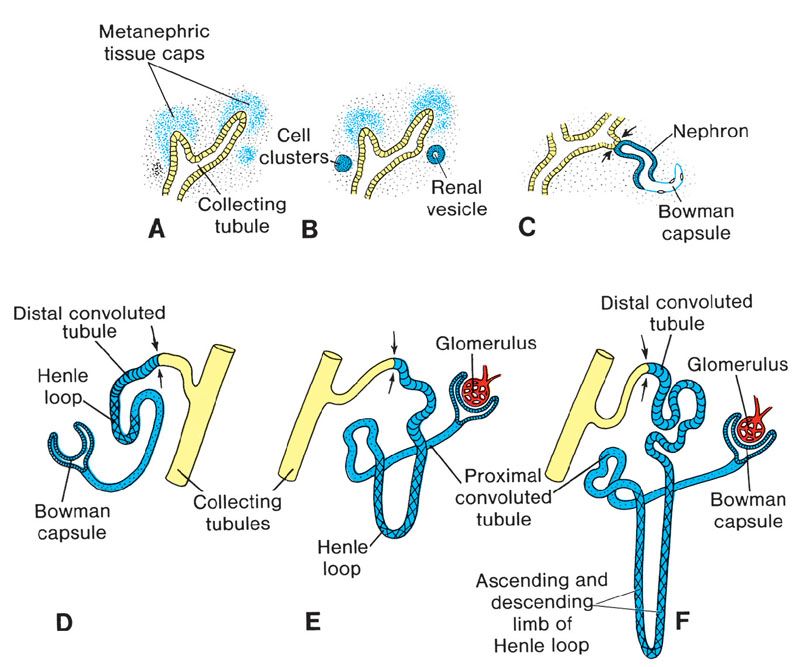
FIGURE 19.1-3: Stages in the development of a metanephric excretory unit (nephron) (A) Metanephric cap stage (B) Renal vesicle stage (C) S-shape tubule stage (D) Bowman’s capsule stage (E) Glomerulus and proximal tubule stage (F) Distal convoluted tubule stage. The arrows point to the place where the excretory unit establishes an open communication that allows a flow of urine from the glomerulus into the collecting system. (From Sadler TW. The urogenital system. In: Sadler TW, ed. Langman’s Medical Embryology. 9th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:321–362.)
The embryo–fetal differentiation of the kidney involves epithelial/mesenchymal interactions under the regulation of many genes, the so-called renal developmental genes (RDG) that include transcription/growth factors and intracellular signaling molecules (e.g., RET, PAX2, HNF1Beta, UMOD, WT1); malfunction or mutation of these genes would induce urinary tract malformation.
The kidneys develop originally within the pelvis and ascend progressively to the lumbar areas (Fig. 19.1-4). The ureteral orifices move cranially into the bladder trigone, whereas the ejaculatory ducts, remains of the mesonephritic ducts, move closer to the midline and enter the prostatic urethra where they open.
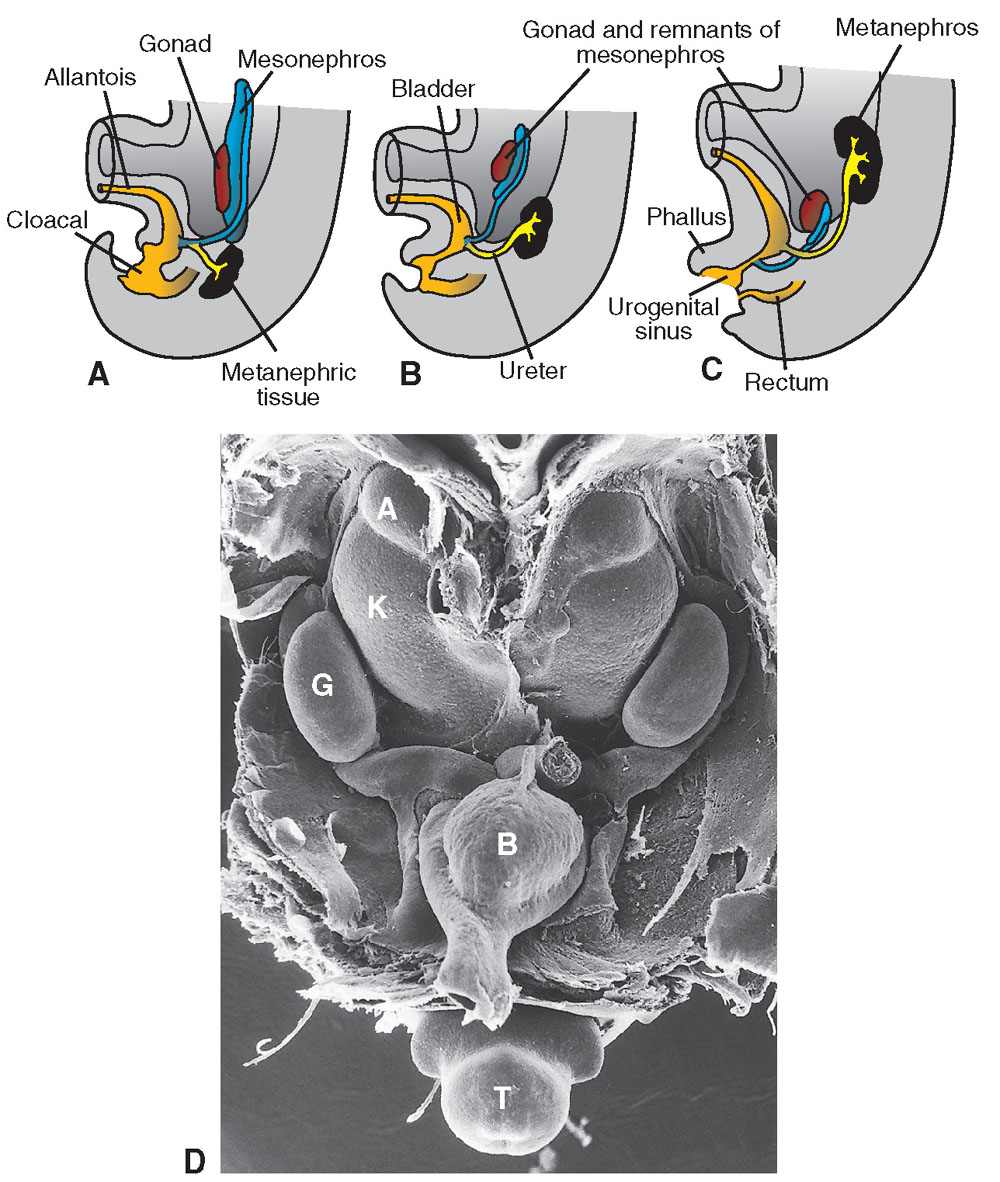
FIGURE 19.1-4: A–C: Cloacal partition into urogenital sinus and bladder anteriorly and rectum posteriorly. The ascent of the kidney is illustrated as well. (D) Scanning electron micrograph of a mouse embryo showing the kidneys in the pelvis. B, bladder; K, kidney; A, adrenal gland; G, gonad; T, tail. In: Sadler TW, ed. Langman’s Medical Embryology. 9th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:321–362.)
The bladder and urethra originate from the cloacal partition by the urorectal septum between the 7th and the 8th week. This partition separates the cloaca into the primitive urogenital sinus anteriorly and the anal canal posteriorly (Fig. 19.1-4). Subsequently, the urogenital sinus develops into three parts: first, the urinary bladder; second, the pelvic part of the urogenital sinus, which gives rise to the urethra (prostatic and membranous in the male, entire urethra in the female) and third, the phallic part that will correspond to the genital tubercle. The anterior urethra in the male develops as an ingrowth of the phallic ectoderm and closure of the urethral folds. Finally, the urogenital membrane breaks during the 7th week, establishing continuity between the developing urinary tract and the amniotic cavity. Chwalla membrane at the ureterovesical junction disappears during the 8th week. Intrinsic narrowings persist and can be discerned at the ureteropelvic and ureterovesical junction up to the 18th week.
If development is normal, urine production starts at the 9th week.8–11,13,14
IMAGING OF THE NORMAL URINARY TRACT
Ultrasound Imaging
The normal development of the urinary tract is well depicted by ultrasound. Bladder and ureter are clearly visible during the first trimester (Fig. 19.1-5). The fetal bladder is the first structure of the urinary tract to be visualized on US, appearing as an anechoic cystic structure within the fetal pelvis around 9 to 10 weeks’ gestation. The kidneys themselves are visualized slightly later, appearing as two hyperechoic oval nodules, one at each side of the lumbar spine. During the first trimester, the kidneys appear globally hyperechoic, without corticomedullary differentiation (CMD). Throughout the rest of the pregnancy, the kidneys will grow and their echogenicity will evolve. The renal growth can be assessed by the measurement of the sagittal length compared with established nomograms (see Table 38 in Appendix A1). Renal length can be evaluated through the formula “renal length in cm = number of gestation weeks × 1.1 inches.” This growth can also be assessed by the evaluation of renal parenchymal area or even volume calculation, although these types of measurements are not performed routinely. With evolving gestational age, the echogenicity of the renal cortex decreases with respect to the liver, and should be less echogenic than the liver by the third trimester. CMD should be progressively demonstrated—hypoechoic medulla compared to relatively hyperechoic cortex—starting at the 4th month of gestation (Figs. 19.1-6A and 19.1-7A). The renal vessels can easily be demonstrated with color or power Doppler (see Fig. 19.1-6B).4,15–19
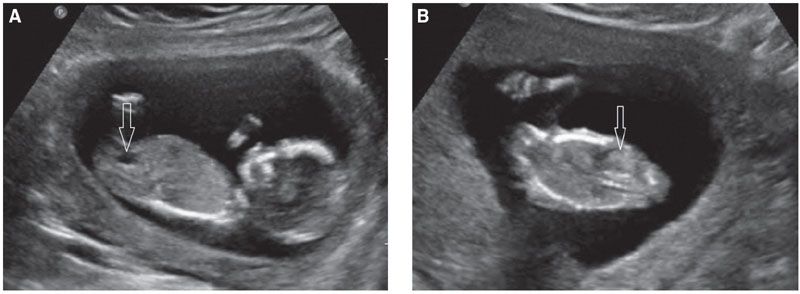
FIGURE 19.1-5: Fetal urinary tract at 13 weeks’ gestation. A: The arrow points to the fluid-filled bladder. B: Coronal view. The arrow points to a hyperechoic normal kidney. Note the hypoechoic adrenal lying above the kidney.
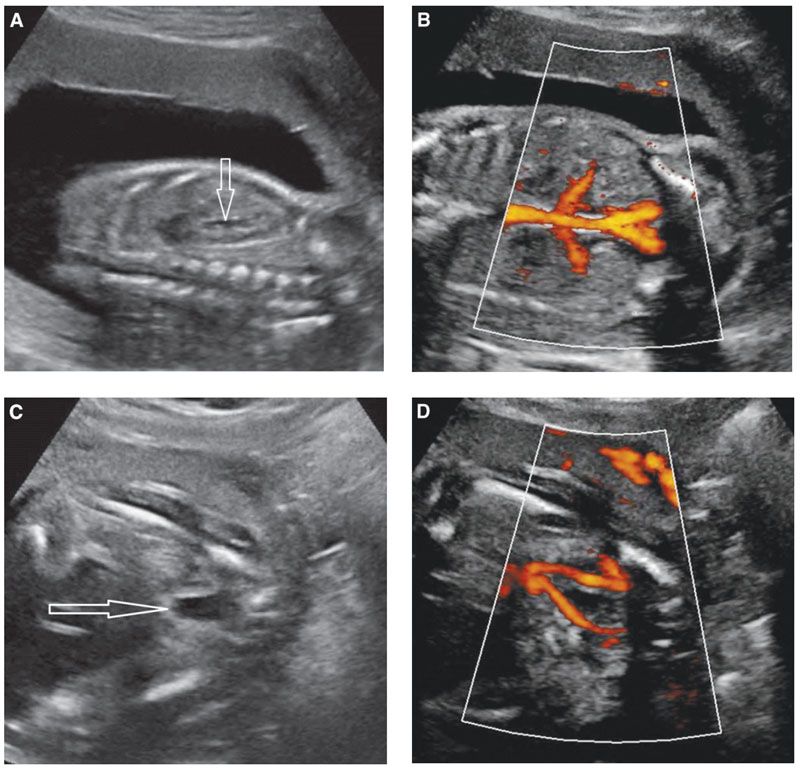
FIGURE 19.1-6: Fetal urinary tract. A: Sagittal ultrasound of a normal kidney in the second trimester. The renal cortical echogenicity has reduced as compared with Figure 19.1-5B. Some urine (arrow) distends the renal pelvis. A corticomedullary differentiation is beginning to appear (hyperechoic cortex/hypoechoic medulla). B: Coronal view of the fetal trunk with power Doppler displaying the aorta and renal vessels. C: Axial view of the lower pelvis and bladder (arrow). D: Color Doppler demonstrates the two umbilical arteries along each side of the bladder.
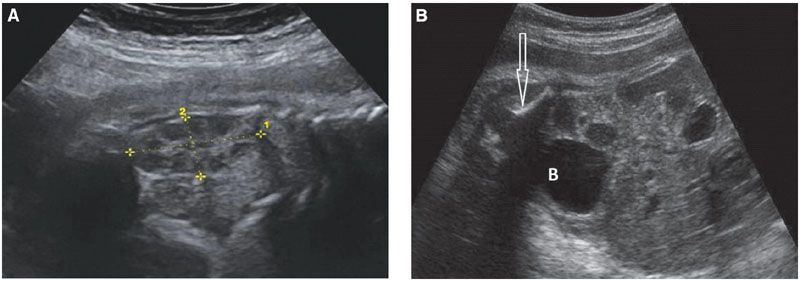
FIGURE 19.1-7: Fetal urinary tract. A: Sagittal view through the left kidney (limited by the crosses) in the third trimester. A corticomedullary differentiation is now obvious (hypoechoic medulla). The kidney measures 36 mm at 32 weeks, appropriate for the calculated length (number of weeks × 1.1 cm). B: Oblique view of the fetal abdomen; a round urine-filled bladder (B) is visible. The arrow points to the iliac wing.
The fetal bladder becomes larger, containing more urine (see Figs. 19.1-6C and 19.1-7B), and cycles of filling and emptying will be observed. These cycles slow at the end of the pregnancy especially in female fetuses. Even almost empty, it should be possible to identify the bladder using the umbilical arteries as landmarks (see Fig. 19.1-6D). Filling of the bladder is influenced by maternal hydration. Normally, the fetal ureters are not visualized.20,21
Amniotic fluid volume should always be assessed. The kidney contributes by two-third to the amniotic fluid volume at the beginning of the 4th month of gestation.22
Information regarding the normal urinary tract can be obtained at each trimester of the pregnancy using US. In the first trimester, a fluid-filled bladder and hyperechoic kidneys should be identified. In the second and third trimesters, a systematic approach to evaluate the urinary tract includes identifying two kidneys, assessing size and echogenicity, including CMD, identifying the bladder, including size and contour, and evaluating the amniotic fluid volume. If an anomaly is identified, it should be determined whether it is isolated or part of a polymalformative syndrome. MRI should be considered if assessment is incomplete.
Magnetic Resonance Imaging
The advantages of MR imaging in the fetus with renal anomalies include a large field of view and multiplanar approach that can help to assess complex and large urinary tract abnormalities and also associated malformations. In cases with oligohydramnios, MR can provide better resolution of anomalies. T2w sequences are well adapted for the visualization of the kidneys, renal pelvis, and bladder. The fetal kidneys display a homogeneous intermediate hypersignal signal as compared with the liver. Urine in the renal pelvis and bladder is high in signal (Fig. 19.1-8). The use of T1w sequences is useful when a vesicourethral fistula is suspected.5,23–28 Diffusion-weighted sequences can be helpful for locating renal parenchyma in cases of possible pelvic kidneys.
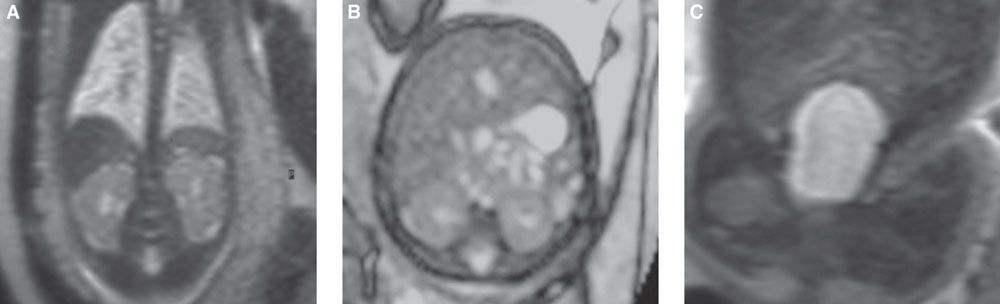
FIGURE 19.1-8: T2w MRI of the normal urinary tract. A: Coronal view through both kidneys, intermediate signal compared with the liver and spleen. B: Axial scan through the kidneys (and liver). C: Coronal view through the bladder.
URINARY TRACT ABNORMALITIES
Bilateral Renal Agenesis
Definition: Congenital absence of both kidneys.
Incidence: Bilateral renal agenesis occurs in 1:4,000 births. The condition is threefold more common in male fetuses, with increased incidence in the offspring of diabetic mothers. It can be isolated or associated with other system malformations and part of syndromes (Table 19.1-3). Bilateral renal agenesis determines the so-called Potter syndrome or sequence (see later).1,9,29–31
Syndromes Including Unilateral or Bilateral Renal Agenesis3
Autosomal Dominant Acro-renal-ocular syndrome Branchio-oto-renal syndrome Ectrodactyly–ectodermal dysplasia—cleft palate (EEC) syndrome Pallister–Hall syndrome Renal–Coloboma syndrome Townes–Brocks syndrome HNF1B (also sporadic) Autosomal Recessive Acro-renal-mandibular syndrome Antley–Bixler syndrome Fraser syndrome Fryns syndrome Rokitansky sequence Smith–Lemli–Opitz syndrome X-linked Goltz–Gorlin syndrome Kallmen syndrome Lenz microphthalmia syndrome Sporadic Caudal regression syndrome Goldenhar syndrome MURCS association Kabuki syndrome Vater syndrome HNF1beta Chromosomal Miller–Dieker syndrome XXX Deletion 22q |
Embryology and Pathogenesis: Renal agenesis can be explained from either failure of the ureteric bud to develop or from absent interaction between the ureteral bud and the metanephritic mesenchyma. Whatever the embryologic “explanation,” it is obvious that mutations of the different genes that are involved in the normal renal development (RET, PAX2, WT1, HNFB1) are responsible for a wide range of anomalies that include uni- or bilateral renal agenesis, (multicystic) dysplasia, or hypoplasia. This explains the recurrence of the disease, sometimes under another phenotype; furthermore, these various genes are also involved in the development of the genital tract, and this explains why genital tract anomalies are associated in the case of their mutations.2,9–11,14,32
Diagnosis
Ultrasound: The sonographic diagnosis of bilateral renal agenesis can be difficult during the first trimester since at this stage, the amniotic fluid volume is normal as it is mainly produced by the placenta; the diagnosis is easier during the second and third trimesters, when it can be based on direct and indirect signs (Fig. 19.1-9). The main direct sign is the lack of visualization of kidneys in the lumbar fossae. Indirect signs include nonvisualization of the bladder (Fig. 19.1-9B), absent renal arteries on color Doppler, oligohydramnios, small chest, and club feet. In most cases, the urine-filled bladder is absent (still, a bladderlike structure can be filled backward through a patent urachus). The combination of renal agenesis, small chest, oligohydramnios, club feet, low-set ears, and facial deformation constitutes the so-called “Potter syndrome” or sequence. Other causes for bilateral nonfunctioning kidneys—e.g., bilateral multicystic dysplastic kidneys—are able to display the same sequence. With bilateral renal agenesis, the adrenals are present and appear elongated (Fig. 19.1-10).32–35
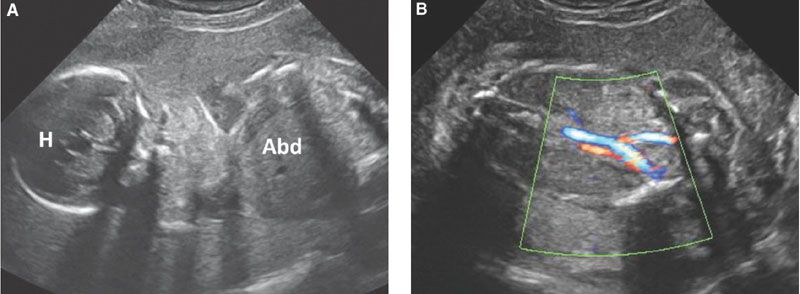
FIGURE 19.1-9: Bilateral renal agenesis. A: Ultrasound through the fetal head (H) and abdomen (Abd) through fetal head (H) and abdomen (Abd). There is a striking anamnios; no renal structure could be demonstrated. B: Color Doppler enhancing the umbilical vessels. No urine-filled bladder is visible. (Courtesy of B. Broussin, MD.)
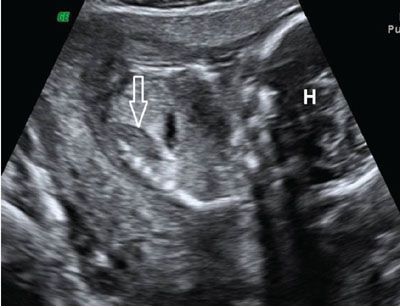
FIGURE 19.1-10: Bilateral renal agenesis. Sagittal ultrasound of the fetal body. A hypertrophied globular adrenal appears as an hypoechoic ovoid mass (arrow). H, fetal head. (Courtesy of R. De Maubeuge, MD.)
MRI: MR imaging is useful to confirm the absence of renal parenchyma and bladder on T2w sequences. Diffusion-weighted sequences may help identify residual renal parenchyma. Associated malformations may be better assessed by MR in the presence of oligohydramnios.5,36
Differential Diagnosis: The differential diagnosis of bilateral renal agenesis includes cases with poorly or nonfunctioning kidneys such as bilateral renal dysplasia or bilateral MCDK (Fig. 19.1-11). Other diagnoses to consider include bilateral pelvic kidneys or a solitary pelvic kidney (Fig. 19.1-12) (see later). Other causes of oligohydramnios such as intrauterine growth retardation or premature rupture of the membranes should also be considered.36,37
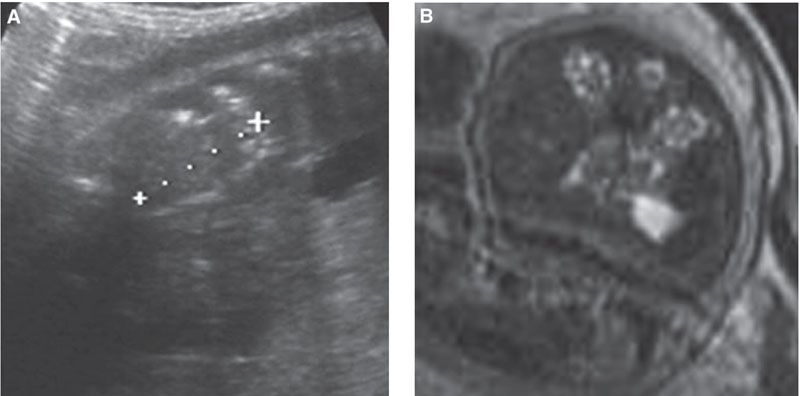
FIGURE 19.1-11: Bilateral multicystic dysplastic kidneys. A: Sagittal ultrasound through one kidney (between crosses), which appears small (20 mm at 30 weeks) and homogeneous, without corticomedullary differentiation. Oligohydramnios is striking. B: T2w MRI. The kidneys have multiple tiny cysts.
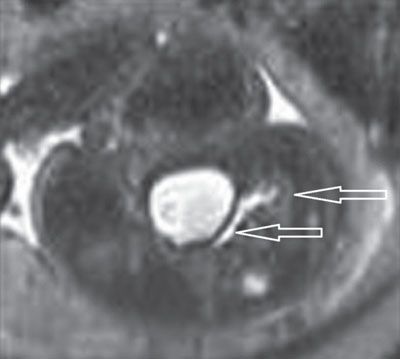
FIGURE 19.1-12: Hypoplastic solitary pelvic kidney. Axial T2w MRI through the fetal pelvis and bladder demonstrates a dilated ureter connected to a hypoplastic kidney (arrows) that was not visualized on simultaneous ultrasound.
Prognosis: Bilateral renal agenesis is lethal.2,36
Management: Genetic counseling is important to evaluate the risk of recurrence.
Chromosomal analysis prenatally may be difficult because of the lack of amniotic fluid.
Recurrence Risk: The recurrence risk ranges between 3% and 6%. There are cases of autosomal dominant and recessive conditions with higher recurrence risks.1–3
Unilateral Renal Agenesis
Definition: Congenital absence of one kidney.
Incidence: Unilateral renal agenesis occurs in about 1:500 to 1:1,000 births. There is a male predominance, and the left kidney is more often the one missing. The condition can be isolated or part of polymalformative syndromes (see Table 19.1-3). The solitary kidney itself can display anomalies such as ureteropelvic junction (UPJ) or ureterovesical junction (UVJ) obstruction as well as vesicoureteric reflux (VUR). Associated genital tract anomalies are commonly encountered.1,9,38
Embryology and Pathogenesis: Renal agenesis can be explained from either failure of the ureteric bud to develop or from absent interaction between the ureteral bud and the metanephritic mesenchyma. Mutations of the different genes that are involved in the normal renal development (RET, PAX2, WT1, HNFB1) are responsible for a wide range of anomalies that include renal agenesis, multicystic dysplasia, and hypoplasia. This explains the recurrence of the disease, sometimes under another phenotype; furthermore, these mutations also explain why genital tract anomalies are associated as some of these genes are also involved in its development.
A pseudorenal agenesis can be the result of a complete involution of a multicystic dysplastic kidney (see later).2,9–11,14,32
Diagnosis
Ultrasound: The sonographic diagnosis of a unilateral renal agenesis is based on the inability to visualize the kidney in one of the renal fossae. The corresponding renal artery tends to be absent on color Doppler (Fig. 19.1-13). The corresponding adrenal fills the empty space, as does the colon with left renal agenesis. Colonic loops should not be misinterpreted as a dilated ureter. Renal abnormalities such as UPJ, UVJ obstruction, or VUR can be associated findings within the solitary kidney. Often, the solitary kidney undergoes compensatory hypertrophy in utero.33,34,38,39
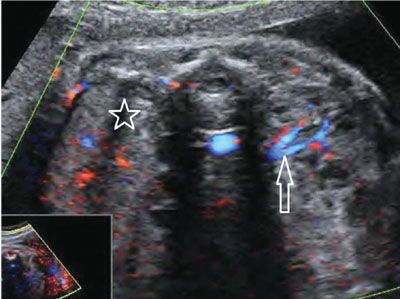
FIGURE 19.1-13: Unilateral renal agenesis. On ultrasound with color Doppler in the second trimester, neither renal parenchyma nor renal vessels can be visualized in the right renal fossa (star), whereas both are visible present on the left side (arrow). (Courtesy of R. De Maubeuge, MD.)
Whenever a renal fossa appears empty, one should look for an ectopic location. Renal agenesis can be the result of a complete involution of a multicystic dysplastic kidney (MCDK).32
MRI: MR imaging is rarely performed for a diagnosis of solitary kidney. T2w and diffusion-weighted images can help search for a potential ectopic kidney. Assessment of other anomalies is useful in polymalformative syndromes (Fig. 19.1-14).5
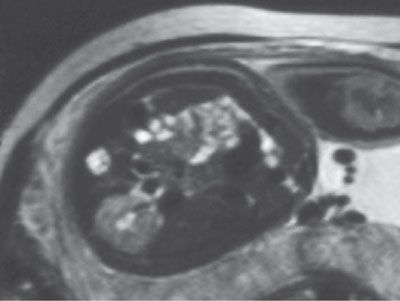
FIGURE 19.1-14: Unilateral renal agenesis. On this T2w MRI, only one kidney is visualized.
Differential Diagnosis: Unilateral renal hypoplasia, crossed fused ectopia, involuting MCDK, pelvic kidney.40
Prognosis: Excellent if isolated. Poor if the remaining kidney is dysplastic or obstructed and poorly functioning or unless the anomaly is part of a severe polymalformative syndrome. Solitary kidneys are at higher risk for developing cardiovascular diseases and renal failure.41,42
Management: Assessment of the remaining kidney is important to exclude UPJ, UVJ, and VUR.
Recurrence Risk: About 12%, possibly under other phenotypes.
Renal Ectopia
Definition: Abnormal location of one or both kidneys. There are different types of ectopia: simple pelvic ectopia corresponds to a kidney that has not migrated from the pelvis, whereas complex ectopia with fusion that includes horseshoe kidneys (HSK) and crossed fused ectopia (CFE). HSK corresponds to kidneys that fuse through an isthmus that connects (most usually) their lower poles. They are usually located lower than the normal kidneys. In CFE, both kidneys are fused on the same side. The ureters open within the bladder trigone in the normal location.43,44
Incidence: The incidence of pelvic kidneys is estimated at 1:2,500, HSK about 1:400 births and CFE around 1:7,500 births. All forms of renal ectopia are associated with a wide range of other renal anomalies, the commonest being UPJ and VUR. There is also a high prevalence of associated other system malformations, most commonly skeletal and genital (approximately 25%).43,44
Embryology and Pathogenesis: Ectopia results from an abnormal ascent of the kidney(s) during embryology. HSK may result from coalescence of the kidney poles during ascent, possibly in relation to abnormal caudal tilting. An abnormal tilting to a higher extent may explain the occurrence of CFE. Vascular and genetic factors are probably involved as well.43,44
Diagnosis: The discovery of an empty renal fossa (or bilateral fossae) may lead to the diagnosis of renal ectopia versus renal agenesis. An ectopic pelvic kidney tends to be small and malrotated. CMD in the renal parenchyma should be searched for (Fig. 19.1-15). The ectopic kidney may have a dilated collecting system, or it could be dysplastic or be multicystic dysplastic (Fig. 19.1-16A).
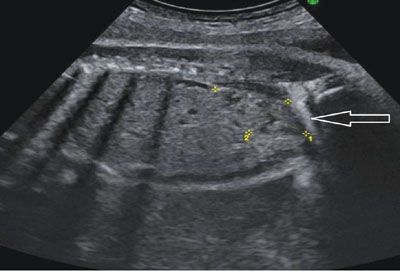
FIGURE 19.1-15: Pelvic kidney. Parasagittal ultrasound of the fetal abdomen shows the pelvic kidney is delineated by the crosses. The arrow points to the right iliac wing. No renal parenchyma could be demonstrated in the left lumbar fossa.
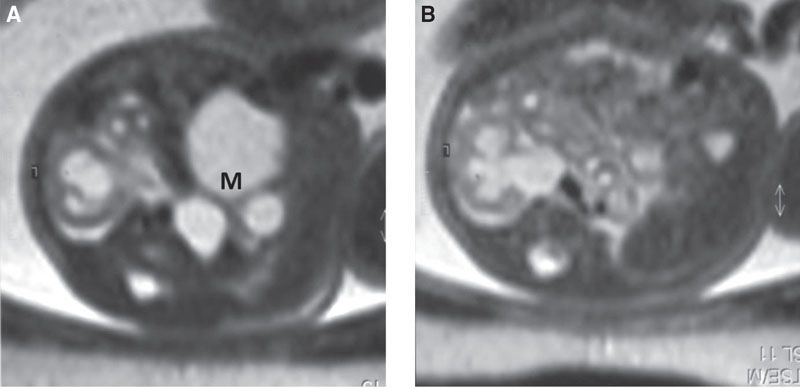
FIGURE 19.1-16: Pelvic MCDK and contralateral UPJ obstruction. A: Axial MRI through the fetal pelvis displays the MCDK (M). B: Axial MRI through the left kidney shows UPJ obstruction.
HSKs may be more difficult to diagnose prenatally since the isthmus can be thin. The axis of the fused kidneys may hint at the diagnosis as the lower poles are more oblique toward the midline than normal. The demonstration of CMD within the renal parenchyma, especially the isthmus, is confirmatory (Fig. 19.1-17). Each kidney can be affected by UPJ obstruction or MCDK.
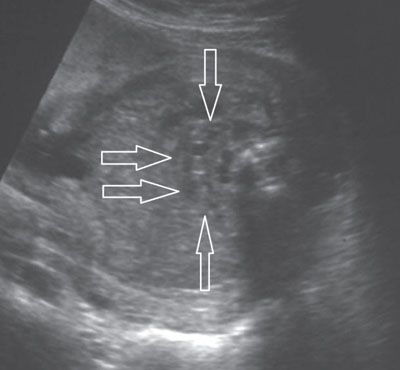
FIGURE 19.1-17: Horseshoe kidney. On axial ultrasound through fetal abdomen in the third trimester, the horseshoe kidney (arrows) can be identified by demonstration of the corticomedullary differentiation.
Cross-fused ectopia is even more difficult to diagnose prenatally. It may be confused with a solitary kidney and/or unilateral renal duplication. The clue to the diagnosis is the axes of both kidneys that can be either parallel to each other or at right angles (L-ectopia). The CFE kidney is typically lower than the normally located one (Fig. 19.1-18).43–46
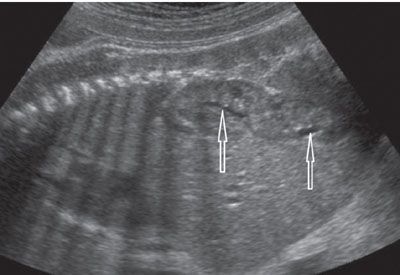
FIGURE 19.1-18: Crossed fused ectopia. On left parasagittal ultrasound in the third trimester, the kidney appears elongated owing to the fusion of both kidneys. Two renal pelves (arrows) are visualized. (Courtesy of C. Garel, MD.)
MR imaging may be useful in evaluating complicated ectopic kidneys, especially those presenting with a multicystic portion or with dilatation.5
Differential Diagnosis: A pelvic kidney may be confused with a nonrenal pelvic mass, such as a pelvic teratoma. CFE can be confused with renal duplication or with solitary kidney.
Prognosis: Ectopic kidneys have a good prognosis unless associated with a significant renal malformation or part of a severe polymalformative syndrome.
Management: There is no specific in utero management. Chromosomal analysis should be considered in the case of polymalformative syndrome.
Recurrence Risk: Around 10% potentially under another phenotype.47
Simple Renal Cyst
Definition: Unilateral, solitary, nongenetically transmitted cystic lesion within the renal parenchyma.51,52,58
Incidence: Rare, most resolving by 24 weeks’ gestation.
Embryology and Pathogenesis: The origin of simple renal cysts is unclear; it may correspond to a sequel of localized ischemia.
Diagnosis
Ultrasound: A simple renal cyst appears as a single cystic structure without septa or calcifications; its size varies from a few millimeters to several centimeters (Fig. 19.1-19). It is localized within the renal cortex. Color Doppler is helpful in order to confirm the intrarenal location of the cyst (Fig. 19.1-19B).
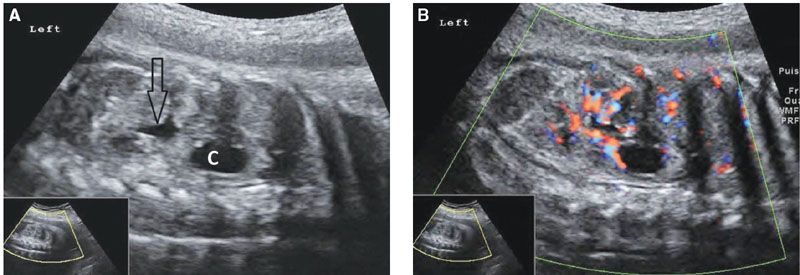
FIGURE 19.1-19: Simple renal cyst. A: On parasagittal ultrasound through the left kidney in the third trimester, a cystic structure (C) is visible within the upper pole of the kidney, and some urine distends the renal pelvis (arrow). B: Color Doppler, same view as in A, confirms the intrarenal location of the cyst. (Courtesy of R. De Maubeuge, MD.)
MRI: At MR imaging, on T2w, a renal cyst will appear as a cystic high-signal structure within the kidney.
Differential Diagnosis: The differential diagnosis includes a calyceal diverticulum, a hydrocalyx, and a cystic tumor. In the case of calyceal diverticulum and hydrocalyx, the diagnosis would be confirmed by the demonstration of a connection with the pyelocalyceal system. A tumor will usually appear as a cystic multiseptated mass. A hereditary renal cystic disease may start with a unilateral cyst; follow-up and familial history would help to reassess the diagnosis. Cysts in the upper pole of a kidney should be differentiated from a dilated upper pole moiety of a duplex kidney.
Prognosis: Excellent if isolated with no specific management in utero.
Cystic Renal Diseases
Definition: Uni- or bilateral presence of multiple cystic lesions within the renal cortex or medulla or both. The cystic lesions may have a genetic (bilateral diseases) or nongenetic transmission (uni- or bilateral diseases). Obstructive cystic dysplasia and multicystic dysplastic kidney (MCDK) are the commonest diseases without genetic transmission. Autosomal recessive (ARPKD) and dominant (ADPKD) polycystic kidney diseases are the commonest ones with genetic transmission. Cystic renal diseases can present as isolated renal anomalies or as part of a syndrome.12,48–50
Embryology and Pathogenesis: The Potter classification of renal cystic diseases has been replaced by a classification based on the genetic or nongenetic origin of the renal cystic diseases. Genetic renal cystic diseases include autosomal dominant and recessive polycystic kidney disease (ARPKD and ADPKD) as well as glomerulocystic kidney disease (GCKD), medullary cystic dysplasia associated with syndromes and nephronophthisis (NPHP)/medullary cystic dysplasia complex.48,49 Among the nongenetic cystic diseases, obstructive cystic dysplasia and MCDK are the most common. Obstructive dysplasia is associated with urinary tract dilatation and various congenital uropathies. Noteworthy, some cases of MCDK are genetically transmitted.
A further step in the characterization and understanding of genetically transmitted cystic renal diseases has been achieved through their recognition as part of the group of ciliopathies (see Table 19.1-2). Primary cilia are microtubules, antennalike cellular organelles that extend outward from the surface of many cells of the renal tubule epithelium. These organelles are rich in receptors, ion channels, and signaling proteins activated by mechanical or chemical stimuli. Cilia regulate cell proliferation and differentiation in the developing and mature kidneys. Any defect in the structures or function of primary cilium may lead to various cystic phenotypes. Cilia are encountered in various sites and organs of the human body (i.e., liver, lungs), and this explains the potential association of cystic renal changes and other malformations encountered in various syndromes.4,15,17 The cilia concept has induced a more global approach in renal cystic diseases combining renal and hepatic diseases. Hepatorenal fibrocystic diseases are characterized by developmental abnormalities of the porto-ciliary hepatic system in association with cystic degeneration of the kidney. They encompass various inherited diseases. The hepatic lesions are usually demonstrated by histology only and to a lesser extent by imaging. One or several genetic defects have been demonstrated (see Table 19.1-2). As some genetic loci mutations can be located close one to another, not surprisingly, different diseases may have similar sonographic appearances (i.e., tuberous sclerosis and ADPKD).49,50
Diagnosis
Ultrasound: Imaging plays an essential role in detecting and characterizing renal cystic diseases as well as follow-up.51–54 Standard high-resolution sonographic images are essential. The knowledge of histologic changes that occur in cystic diseases helps one to understand their sonographic appearance (Table 19.1-4).55 The exam includes measuring renal size, defining echogenicity of the cortex compared with liver and medulla (hypoechoic, absent, or hyperechoic), assessment of the CMD (present, absent, or reversed) and presence of cysts (number, size, and location), or calcifications as well as evaluation of the pelvicalyceal system.

Two main sonographic features lead to the suspicion of renal cystic diseases: the presence of renal cysts and the detection of hyperechoic kidneys (Fig. 19.1-20).51,52,57 The “cysts” that are visualized on US result either from renal tubular dilatation, glomerular cysts, or from real cysts. They develop anywhere in the kidney and can be uni- or bilateral. Renal hyperechogenicity may involve the entire kidney, only the cortex, or only the medulla. The discovery of hyperechoic kidneys can be challenging as there are no objective US criteria defined up to 32 weeks’ gestation; after 32 weeks, the hyperechogenicity is less subjective and should be considered abnormal whenever the renal cortex appears more echoic than the liver or spleen.4 Once a suspicion of a renal cystic disease has arisen, the evaluation of the US findings should be standardized in a decision tree-type approach (Table 19.1-5).51
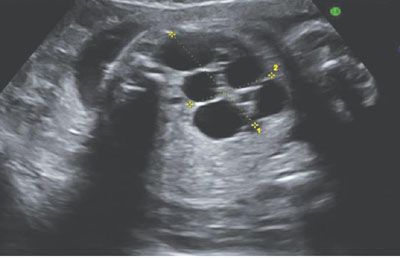
FIGURE 19.1-20: Left multicystic dysplastic kidney. Ultrasound in the second trimester shows typical multicystic patter, and no renal parenchyma or renal pelvis can be visualized.
Differential Diagnosis of Fetal Hyperechoic and Cystic Kidneys
Hyperechoic Kidneys with Normal or Moderately Enlarged Kidneys Renal cystic diseases TCF2 gene mutation Autosomal recessive polycystic kidney disease Autosomal dominant polycystic kidney disease Maternally induced diseases Diabetes Neutral endopeptidase autoimmune glomerulopathy Cytomegalovirus infection Antagonists Angiotensin II Metabolic diseases Nephrotic syndromes, Finnish type Obstructive dysplasia “Transient” Bilateral Multiple Cysts Autosomal recessive polycystic kidney disease Autosomal dominant polycystic kidney disease Bilateral multicystic dysplastic kidney disease (only in fetus) Bilateral obstructive dysplasia Syndromes (e.g., Bardet–Biedl, Zellweger) Fetal Macrocysts (>2 cm) Tuberous sclerosis Simpson–Golabi–Behmel syndrome TCF2-gene mutation Joubert syndrome |
Color Doppler assessment is useful in selected cases.9 Associated malformations should be searched for with particular assessment of the liver and genital tract. Family history is important, and whenever cystic kidneys are suspected, it is useful to perform a sonographic examination to the parents to check for “unknown” familial disease.
MRI: In some patients, MR imaging provides additional information by demonstrating the renal cysts or associated brain malformation.5,56
Management: When bilateral renal cystic disease is identified, the evaluation of the US findings can be standardized in a decision tree-type approach. The first step is to rule out dysplasia associated with an obstructive uropathy (with dilatation of the collecting system). Obstructive dysplasia is the commonest cause for a hyperechoic cortex with or without cysts. MCDK is the second diagnosis to consider. If these diagnoses are unlikely, inherited renal cystic diseases should be considered.
Knowledge of any familial history is essential. If positive, the finding of abnormal kidneys suggests recurrence (Fig. 19.1-21). If no familial history is present, cases can be further separated in patients where the renal findings are isolated and limited to the kidney and those where the renal anomalies are associated with malformations of other organs.
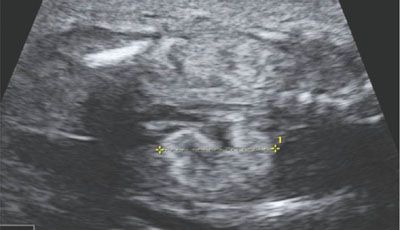
FIGURE 19.1-21: Glomerulocystic disease. Coronal ultrasound of both kidneys, which appear hyperechoic but normal sized. Family history was positive. (Reproduced from Avni FE, Garel C, Cassart M, et al. Imaging and classification of congenital cystic renal diseases. AJR Am J Roentgenol. 2012;198:1004–1013, with permission.)
There are typical US patterns suggestive of specific diagnoses and nontypical US patterns. A “typical” pattern means typical pattern of all sonographic features that lead to a specific diagnosis (isolated or in association with other malformations). Nonspecific (or nontypical) means that there are no significant features that can lead to a specific diagnosis. For the latter, the diagnosis can be approached using tables of differential diagnosis (Fig. 19.1-22) (Table 19.1-6). In undetermined cases, supplementary examinations may help, such as genetic studies or MR to reach the most accurate diagnosis.
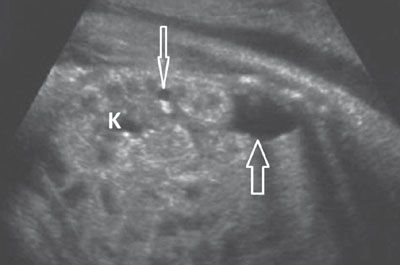
FIGURE 19.1-22: Simpson–Golarbi–Behmel syndrome with renal cysts. Sagittal ultrasound shows a kidney (K) that appears hyperechoic and contains one large and one tiny (arrows) cyst. Small cysts were visualized in the other kidney as well. Anomalies included signs of an overgrowth syndrome.
Syndromes that Include Cystic Kidneys3
Bardet–Biedl syndrome COACH syndrome Ellis–Van Creveld syndrome Fryns syndrome Glutaric acid II Jeune syndrome Marden–Walker syndrome Joubert syndrome Meckel–Gruber syndrome Oro-facio-digital syndrome Roberts syndrome Smith–Lemli–Opitz syndrome Zellweger syndrome Tuberous sclerosis Trisomy 13 Deletion 22q |
For example, in the fetus, bilateral, very large (>4 SD) hyperechoic kidneys with hyperechoic medulla (reversed CMD) is a typical pattern for fetal ARPKD (Fig. 19.1-23).The association of “renal cystic changes + malformations” can be characteristic for a specific syndrome (see Table 19.1-6). For example, GLMCK + Molar tooth sign in the CNS establishes the diagnosis of JOUBERT syndrome (Fig. 19.1-24).57,51–53
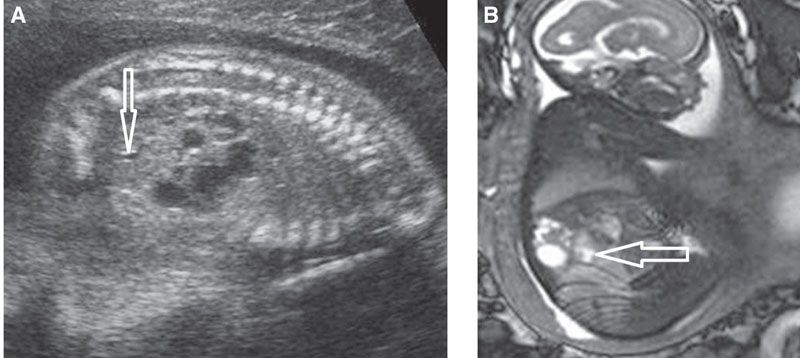
FIGURE 19.1-23: MCDK developing within the upper poles of a duplex kidney bilaterally. A: Parasagittal US of one kidney demonstrates multicystic appearance of the upper moiety. The lower pole pelvis is slightly dilated (arrow). B: Parasagittal MRI confirms the lower moiety is displaced anteriorly and slightly distended (arrow).
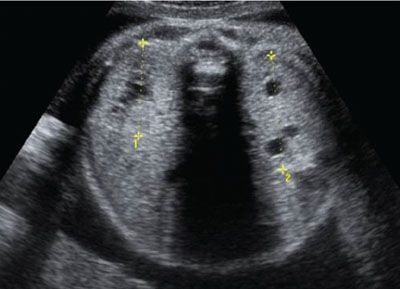
FIGURE 19.1-24: Joubert syndrome. Axial ultrasound shows both kidneys to be hyperechoic without CMD. There are scattered cysts within the parenchyma around the CM junction. The fetus displayed a molar tooth sign on fetal MR imaging (not shown). (Courtesy of M. Molho, MD.)
Cystic Obstructive Dysplasia
See section on Urinary Tract Dilatation.
Multicystic Dysplastic Kidney Disease
Definition: Multiple cysts of varying size that do not connect to a central pelvis. No functioning renal parenchyma.39,40,59–63
Incidence: Unilateral MCDK occurs in 1:2,500 births, bilateral MCDK occurs in 1:12,000 births.
Embryology and Pathogenesis: MCDK is classically considered to result from aberrant interactions between the ureteral bud and the metanephritic blastema. Most cases are sporadic, yet a genetic origin has been established in some cases. Mutations of PAX2, TCF2 (HNF1β), and uroplakin have been identified.
On histology, the kidney is highly dysplastic, displaying cysts, primitive abnormal tubules, few abnormal nephrons, and foci of cartilage. In rare instances, a few functioning normal nephrons are present, and the kidney would display some function. It can be uni- or bilateral (lethal form). It can develop in ectopic or duplex kidneys. Unilateral MCDK is associated with contralateral renal anomalies such as UPJ or VUR in 30% to 40% of cases. It can be associated with malformations of several other systems, especially of the genital tract.
Ultrasound: MCDK is usually detected during the second-trimester US examination. Its diagnosis is straightforward in typical cases: multiple cysts of varying sizes without normal renal parenchyma and without recognized renal pelvis (Fig. 19.1-20). The cysts may be tiny (microcystic form) (Fig. 19.1-25) or very large (giant form) (Fig. 19.1-26). The homolateral ureter may be enlarged. Furthermore, a MCDK may affect an ectopic kidney (see Fig. 19.1-16A) or any moiety of a duplex one (Fig. 19.1-27). Once a MCDK has been detected, associated anomalies should be searched at the level of the contralateral kidney (see Fig. 19.1-16B), the genital tract, or other systems. The contralateral kidney may undergo compensatory hypertrophy already in utero, which can be assessed through measurement of the contralateral kidney (see Fig. 19.1-25).39 Furthermore, MCDK may undergo regression in utero or after birth, resulting eventually in a pseudorenal agenesis.40,64
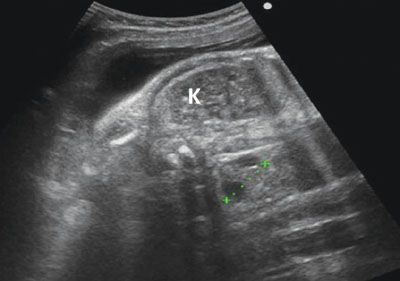
FIGURE 19.1-25: MCDK with contralateral hypertrophy. Oblique ultrasound in the third trimester shows the small MCDK (limited by the crosses), which is displaying small cysts. The contralateral kidney (K) measures 42 mm in length at 30 weeks, which confirms hypertrophy.
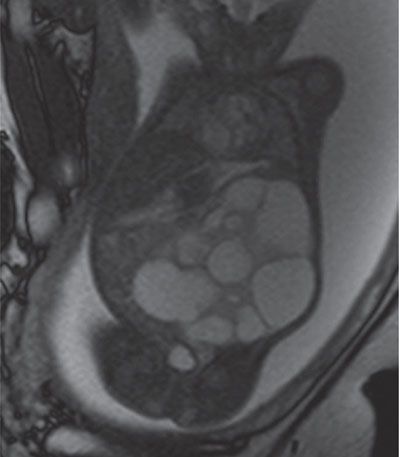
FIGURE 19.1-26: MCDK. T2w coronal MRI shows a large MCDK occupying a large part of the fetal abdomen. (Courtesy of K. Chaumoitre, MD.)
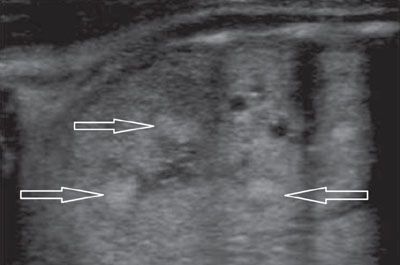
FIGURE 19.1-27: ARPKD with hyperechoic medulla. Sagittal US of one kidney (both displayed the same pattern) shows areas of mottled hyperechogenicities within the renal pyramids (arrows). (Courtesy of C. Garel, MD.)
MRI: MR imaging may be helpful to characterize very large or ectopic MCDK and unusual complications of the anomaly as well as complications of the contralateral kidney (see Figs. 19.1-16, 19.1-23, and 19.1-27
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


