System
Adverse effects
Metabolic/endocrine
Weight gain/obesity, fluid retention/edema, hypokalemia, diabetes mellitus/insulin resistance, adrenal insufficiency, amenorrhea
Dermatologic
Skin thinning, purpura, Cushingoid appearance, acne, hirsutism
Ophthalmologic
Posterior subcapsular cataract, glaucoma, central serous chorioretinopathy
Cardiovascular
Arrhythmia, hypertension, dyslipidemia, premature atherosclerotic disease
Gastrointestinal
Peptic ulcer disease (especially, concurrent user of NSAIDs), acute pancreatitis, steatohepatitis, visceral perforation
Musculoskeletal
Osteoporosis, osteonecrosis, myopathy, growth retardation
Neuropsychiatric
Mood disorder: depression, mania, anxiety, and emotional irritability; psychosis; insomnia
Immune
Increased risk of infections: bacterial, fungal, parasitic, and viral; reactivation of latent viruses
11.2 Prevalence
GI-FHON may develop in patients receiving glucocorticoids in short-term high doses or in long-term doses and even after intra-articular injection or application of topical preparations. Since glucocorticoids have been widely used as therapeutic drugs to treat numerous diseases in various regimens, the reported prevalences of GI-FHON differ among investigators. Most reports have documented the prevalences in patients with systemic lupus erythematosus (SLE), renal transplantation, or leukemia. In prospective studies where 50 or more patients were included and an MRI was used for diagnosis [5–23], 14.5–34.6 % of SLE patients were diagnosed as having GI-FHON (Table 11.2). Of patients undergoing kidney transplantation, 6.0–24.7 % were reported to suffer from GI-FHON. On the other hand, GI-FHON occurred in 4.3–11.3 % of patients with hematologic diseases, including acute lymphoblastic leukemia.
Table 11.2
Prevalence of glucocorticoid-induced femoral head osteonecrosis reported in prospective studies
Authors | Publication year | Underlying disease | Patients | Age | Follow-up period | Prevalence | Reference | |
|---|---|---|---|---|---|---|---|---|
FHON | Total ON | |||||||
Zizic et al. | 1985 | SLE | 54 | 18–67 | 51.9 % | [5] | ||
Ono et al. | 1992 | SLE | 62 | Mean 30.7 | ≤5 years | 14.5 % | [6] | |
Sugano et al. | 1994 | SLE | 60 | 14–57 | 3–7 years | 15.0 % | [7] | |
Oinuma et al. | 2001 | SLE | 72 | 13–66 | ≤12 months | 31.9 % | 44.4 % | [8] |
Nakamura et al. | 2010 | SLE | 169 | 34.6 % | 38.5 % | [9] | ||
Shigemura et al. | 2011 | SLE | 173 | 1 year | 37.1 % | [10] | ||
Kopecky et al. | 1991 | Kidney TPL | 97 | 14–59 | 24 months | 10.3 % | [11] | |
Tervonen et al. | 1992 | Kidney TPL | 100 | 24–75 | 0.5–25.5 years | 6.0 % | [12] | |
Marston et al. | 2003 | Solid organ TPL | 52 | 24–65 | 0.3–4.3 years | 13.5 % | [13] | |
Shibatani et al. | 2008 | Kidney TPL | 150 | 16–36 | ≤12 months | 24.7 % | [14] | |
Guichelaar et al. | 2007 | Liver TPL | 360 | ≤8 years | 6.1 % | 7.5 % | [15] | |
Ribeiro et al. | 2001 | ALL, NHL | 116 | 7.8 % | 14.7 % | [16] | ||
Tauchmanovà et al. | 2003 | Stem cell TPL | 207 | 18–59 | 5.8 % | [17] | ||
Talamo et al. | 2005 | MM | 553 | 25–77 | 5–114 months | 8.9 % | [18] | |
Schulte et al. | 2005 | Stem cell TPL | 255 | 15–59 | >5 years | 4.3 % | [19] | |
Niinimäki et al. | 2007 | ALL | 97 | 1.2–15.3 | 11.3 % | 23.7 % | [20] | |
te Winkel et al. | 2011 | ALL | 574 | Mean 6.4 | 2.5–3.3 months | 4.4 % | 6.6 % | [21] |
Kawedia et al. | 2011 | ALL | 194 | 45.9 % | [22] | |||
Wing et al. | 1998 | Spinal cord injury | 59 | 15–64 | 6–46 months | 0.0 % | 0.0 % | [23] |
Shigemura et al. | 2011 | Inflammatory diseases | 129 | 1 year | 20.9 % | [10] | ||
11.3 Risk Factors
Because GI-FHON develops in patients with different underlying conditions, risk factors are not homogeneous in all disease states.
11.3.1 Risk Factors for GI-FHON in Animal Models
The dose of the glucocorticoids has been the primary focus of research on risk factors for GI-FHON. Many human studies have described associations between glucocorticoid doses and the prevalences of GI-FHON, while others have not. For example, a study analyzing 24 cohorts receiving glucocorticoids reported a 4.6 % increase in GI-FHON development as the doses of oral prednisone increase by 10 mg/day [24]. Animal models have shown that higher doses are more likely to develop GI-FHON [25]. It has been reported that rabbits are more susceptible to the development of GI-FHON when treated with methylprednisolone than to prednisolone or triamcinolone in [26], although there is no similar evidence in humans. Among rabbits receiving methylprednisolone, those with increased low-density lipoprotein (LDL) cholesterol to high-density lipoprotein (HDL) cholesterol, increased free fatty acids, or decreased hepatic cytochrome P4503A activity are at high risk of developing GI-FHON [27, 28].
11.3.2 Risk Factors in SLE Patients
In SLE patients, a daily prednisone dose of >30–40 mg has been reported to be associated with GI-FHON [6, 29]. However, cumulative or the highest dose has been heterogeneously reported as a risk factor by different investigators. Additionally, methylprednisolone pulse therapy and cytotoxic agents have also been found to be related to GI-FHON [29–32]. Generally, SLE patients with more active diseases are more likely to receive glucocorticoids in higher doses of methylprednisolone pulse therapy or cytotoxic agents. However, there are discrepancies regarding results on SLE disease activity between patients with and without the development of GI-FHON [33, 34]. The majority of GI-FHON cases develop within 3–12 months of initiation of glucocorticoid treatment [8, 31]. Increases in glucocorticoid doses secondary to a relapse of SLE may be associated with GI-FHON [34].
In addition to the use of glucocorticoids, several features of SLE have been identified as risk factors: thrombophlebitis, vasculitis, Raynaud’s phenomenon, renal dysfunction, arthritis, smoking, preeclampsia, and so on [6, 30]. In contrast, nested case-control studies have revealed that the use of glucocorticoids is the only risk factor for GI-FHON [32, 33]. Studies on the association between antiphospholipid antibodies and GI-FHON have reported different results [35–37]. Therefore, a specific manifestation may not be a risk factor for GI-FHON in SLE patients.
Because not all SLE patients do develop GI-FHON when receiving high-dose glucocorticoids, the presence of additional regional risk factors or individual variations of glucocorticoid sensitivity can be involved in the occurrence of GI-FHON. For example, age at the time of initial glucocorticoid administration can affect the development of GI-FHON. A prospective study has revealed that GI-FHON did not develop in SLE patients at the age of <14 years, suggesting that children may tolerate ischemia due to their abundant vascularity with growth plates and red marrow [9]. In addition, posttreatment presentation of Cushingoid body phenotype, which is a characteristic feature of glucocorticoid excess and does not develop in patients with glucocorticoid resistance, is reported to be a risk factor [29, 30, 35].
11.3.3 Risk Factors in Kidney Transplant Patients
Kidney transplantation is the treatment of choice for patients with end-stage renal diseases. Just after transplantation, immunosuppressive therapy, including glucocorticoids, is needed to prevent and treat acute graft rejection and to avoid chronic graft damage. It has been shown that the risk of GI-FHON is associated with glucocorticoid doses [14, 38, 39]. In particular, the total dose of glucocorticoids during the first 7 months can have a significant risk [14, 38]. Introduction of immunosuppressants, such as cyclosporine or tacrolimus, can decrease the incidence of GI-FHON in kidney transplant patients [38] because these immunosuppressants reduce glucocorticoids doses. An association of the incidence of GI-FHON with a history of acute rejection is not consistently reported [14, 39].
11.3.4 Risk Factors in Patients with Hematologic Malignancies
Glucocorticoids successfully treat acute lymphoblastic leukemia (ALL), lymphoma (NHL), and multiple myeloma (MM) because they can kill hematologic malignant cells. Thus, many treatment regimens include glucocorticoids, such as dexamethasone. Since the prevalence of ALL is higher in children than in adults, most GI-FHON studies in ALL patients have been conducted on pediatric patients. ALL patients at the age of >10 years increase the risk of GI-FHON [16, 20–22, 43, 44], and old age is also an independent risk factor in adult patients with MM [18]. Additionally, female patients have a higher risk of GI-FHON than males [20, 21, 43, 44]. Intensive therapy with dexamethasone confers a high incidence of GI-FHON [20, 22], and alternate-week scheduling of dexamethasone can reduce the development of GI-FHON [43, 44]. In some studies, patients receiving dexamethasone more frequently developed FHON than those receiving prednisone [43]. An association between FHON and poor dexamethasone clearance has been reported [22]. In patients undergoing stem cell transplantation, both an episode of graft-versus-host disease (GVHD) and the use of glucocorticoid are risk factors [17]. The incidence of GI-FHON has been reported to be higher in patients receiving unrelated donor transplants than in those receiving allogenic matched related donor or autologous transplants [17, 45].
11.4 Pathogenic Mechanisms
The pathophysiology of GI-FHON is multifactorial, complex, and poorly understood. Although host factors and underlying diseases have been shown to play a significant role in the development of GI-FHON, investigators have failed to explain why only a fraction of patients are at greater risk than others. Additionally, the multisystemic effects of glucocorticoids and their interactions make the pathological mechanisms more complicated. In this context, the multi-hit theory proposed by several investigators is a plausible explanation for the development of GI-FHON [46, 56]. In susceptible patients who have a genetic predisposition or an underlying disease that threatens bone and vascular tissues, the accumulative glucocorticoid effects may result in the occurrence of GI-FHON (Fig. 11.1). Genetic factors will be discussed in another chapter.
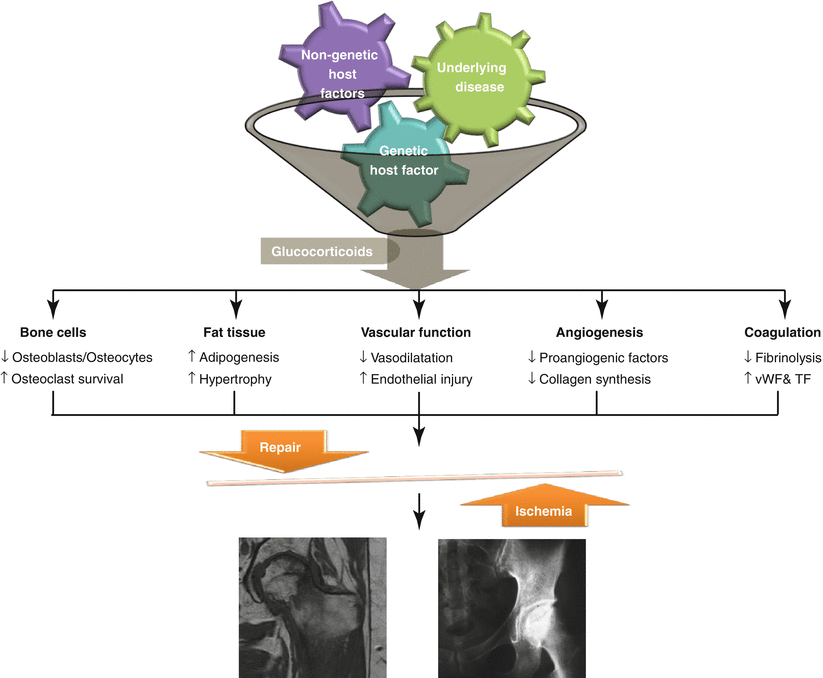
Fig. 11.1
Schematic presentation of the multifactorial pathogenesis of glucocorticoid-induced femur neck osteoporosis. vWF von Willebrand factor, TF tissue factor
11.4.1 Effects of Glucocorticoids on Bone Cells
Animal and human studies have shown that glucocorticoids can induce apoptosis of osteoblasts and suppress the production of osteoblasts in the bone marrow [47]. Osteoblast-/osteocyte-specific 11β-hydroxysteroid dehydrogenase type 2 (HSD2, the enzyme metabolizes glucocorticoids into inactive metabolites) transgenic mice protect from prednisolone-induced decrease in osteoblast survival, osteoblast number, and bone formation [48]. On the other hand, the survival of osteoclasts can be prolonged by glucocorticoids [49]. The interaction of receptor activator of NF-κB ligand (RANKL) and osteoprotegerin (OPG) is a major determinant of osteoclastogenesis. RANKL and macrophage colony-stimulating factor (M-CSF) are essential for osteoclastogenesis, while OPG prevents RANKL from binding to RANK, resulting in inhibition of osteoclastogenesis. Glucocorticoids increase RANKL and M-CSF expression and decrease OPG expression by human osteoblastic and stromal cells in culture [50, 51]. From the results of studies using mice with conditional osteoclast-specific deletion of glucocorticoid receptors [52], glucocorticoids inhibit proliferation of osteoclast precursors and mature osteoclast-mediated bone resorption. Such glucocorticoid-induced suppression of osteoclasts induces blunting of osteoblast function in the context of osteoclast-osteoblast interactions throughout bone remodeling [53]. Also, glucocorticoids suppress the secretion of sex hormones and the expression of bone morphogenetic protein-2, insulin growth factor 1, and osteocalcin [54, 55]. Furthermore, calcium absorption from the kidney and intestine is disturbed by glucocorticoids [54].
Osteocytes are the most abundant cells (>95 %) in the bone and embedded in mineralized bone matrices. They are thought to play a pivotal role as mechanosensors and initiators of the bone remodeling process [56]. Living osteocytes directly stimulate osteoblastogenesis and inhibit osteoclastic resorption through OPG expression. The femurs from patients with GI-FHON show many apoptotic osteocytes, anatomically juxtaposed to the osteonecrotic fractures [57]. Glucocorticoid-induced osteocyte apoptosis results in the mechanosensory dysfunction of the osteocyte network and consequently leads to impairment of bone repair processes. Additionally, hypoxia-inducible factor (HIF)-1α, a critical regulator of cellular response in hypoxic condition, can alter the mechanosensitivity of osteocytes and suppress load-induced bone formation [58].
11.4.2 Effects of Glucocorticoids on Fat Tissue
Glucocorticoids promote differentiation of preadipocytes to mature adipocytes via upregulation of the peroxisome proliferator-activated receptor (PPAR)-γ and downregulation of runt-related transcription factor 2 (Runx2). In addition, glucocorticoids can cause adipocyte hypertrophy through increased synthesis and storage of lipids [59]. It has been shown that the number and area of fat cells increase in the bone marrow after exposure to glucocorticoids [60, 61]. These changes can generate compression of venous sinusoids and congestion in the bone marrow. As a result, adequate arterial blood flow may not be achieved due to increases in intraosseous pressures, eventually leading to osteonecrosis [62].
11.4.3 Effects of Glucocorticoids on Vascular Functions
Patients with Cushing syndrome have been reported to have increased cardiovascular morbidity and mortality. Chronic administration of glucocorticoids inhibits the synthesis of vasorelaxants, such as prostaglandin E1, prostacyclin, and endothelial nitric oxide (NO), via a suppression phospholipase A2 and endothelial nitric oxide synthase (eNOS) [63]. In addition, glucocorticoids indirectly increase vascular tone through the upregulation of angiotensin II type I receptor and α-1 adrenergic receptors in vascular smooth muscle cells [64]. Furthermore, glucocorticoids can stimulate the synthesis of vasoconstrictor endothelin-1 and potentiate its vasoconstricting effect in vascular smooth muscle cells [65]. There is some evidence that glucocorticoids increase reactive oxidative stress species (ROS) which decrease NO availability [66]. Thus, glucocorticoids can cause dysregulation of endothelium-dependent and endothelium-independent vasodilatation. Furthermore, higher doses of dexamethasone were reported to result in microvascular endothelial cell apoptosis and could lead to capillary rarefaction in a glucocorticoid-induced hypertension model [67]. This may be involved in glucocorticoid-induced hypertension and hypercoagulability. The effect of glucocorticoids on the coagulation system is discussed below.
11.4.4 Effects of Glucocorticoids on Angiogenesis
New blood vessel formation is essential for the repair of ischemia-damaged tissues when FHON develops. However, glucocorticoids have been shown to inhibit angiogenesis. In a dexamethasone-induced FHON rabbit model, vascular endothelial growth factor (VEGF) gene therapy improved the repair process of osteonecrosis [68]. Glucocorticoids suppress expression of VEGF and increase expression of antiangiogenic thrombospondin-1 [69, 70]. Glucocorticoid-induced suppression of matrix metalloproteinases and plasminogen activators may affect the proangiogenic process through impairment of basement membrane turnover [71]. It has also been shown that GCs can inhibit capillary growth by reducing collagen synthesis by myofibroblasts [72]. Because cartilage components released from subchondral fracture sites have been considered antiangiogenic, the negative effects of glucocorticoids on angiogenesis may be potentiated in the subchondral bone tissue in FHON cases [73].
11.4.5 Effects of Glucocorticoids on the Coagulation System
Fibrinolytic activity is balanced by tissue plasminogen activator (t-PA) and plasma plasminogen activator inhibitor-1 (PAI-1). The accumulated evidence of in vitro studies as well as studies of patients with Cushing syndrome suggests that glucocorticoids increase the PAI-1 activity, resulting in a relatively hypercoagulable state [74]. The plasma level of the von Willebrand factor (vWF) is a marker of endothelial cell damage because the vWF is synthesized and stored in the endothelial cells. Plasma vWF levels are reported to increase in subjects with exogenous or endogenous excess glucocorticoids, and dexamethasone induces the expression of vWF, cell adhesion molecules, and tissue factor in vascular endothelial cells [75]. Since vWF is involved in platelet aggregation and adhesion, glucocorticoid-induced endothelial damage may contribute to thrombi formation.
11.5 Clinical Manifestations
The clinical features of GI-FHON are not different from those in other FHON. Patients with GI-FHON usually experience pain primarily in the groin but occasionally the buttocks. The pain onset often can be described as acute. The pain is usually deep and throbbing and becomes worse with ambulation. Patients frequently complain of a catching or popping sensation with motion. In patients with a history of glucocorticoid treatment, especially in higher doses, persistent hip pain with joint movement, tenderness, or reduced range of motions indicates the need for a prompt workup for GI-FHON. On physical examination, limitation of internal rotation in both flexion and extension is observed, with passive internal rotation in extension being particularly painful. A Trendelenburg gait is often present. Additionally, some characteristic features of chronic glucocorticoid users can be seen: moon face, central obesity, and buffalo hump.
Anteroposterior radiographs and frog lateral radiographs of both hips are the primary diagnostic modalities, while plain radiographic findings are frequently normal. Magnetic resonance imaging (MRI) is ideal if x-ray findings are normal and clinical suspicion is high [9, 37]. The sensitivity and specificity of MRI are both greater than 98 % for the diagnosis of osteonecrosis, which is higher than those of other diagnostic modalities. Therefore, MRI should be performed in all patients with osteonecrosis to assess the extent of the disease. Three-dimensional MRI with image registration can be used to assess changes in lesion size [76, 88]. Bone scan may be helpful when x-ray findings are normal and when MRI cannot be performed. It may be low-cost alternative when the suspicion index is low [77, 89].
11.6 Natural Course
Most studies on the natural history of FHON have been conducted on patients with heterogeneous subtypes of nontraumatic FHON. However, the natural history of nontraumatic FHON cannot represent that of GI-FHON because subtypes of nontraumatic FHON may have different pathogenic mechanisms and underlying conditions necessitating glucocorticoids can affect the course of FHON. The results of studies using MRI to diagnose asymptomatic GI-FHON are summarized in Table 11.3 [7, 11, 78–83]. About one-third of patients with asymptomatic early GI-FHON have symptomatic or radiological progression over a period of several years. Thus, clinical progression or subchondral collapse rate seems to be lower than the other subtypes of nontraumatic FHON, especially FHON associated with sickle cell anemia. Sometimes small lesions of GI-FHON are reported to be spontaneously resolved [81–83]. However, as with other subtypes, femoral head collapse develops most often in GI-FHON patients with larger osteonecrosis areas (>15–30 %), lesions occupying the weight-bearing surface, higher radiographic stages, or hip pain [84]. There is no established risk stratification system to exactly estimate outcomes in GI-FHON patients. Because proper therapeutic decisions depend on the natural course of FHON, more extended studies are needed to develop an improved risk classification system.
Table 11.3
Progression of asymptomatic GI-FHON
Authors | Publication year | Underlying disease | Hip no. | Initial status | Follow-up period | Progression | Reference | ||
|---|---|---|---|---|---|---|---|---|---|
Mean | Range | Symptom | Collapse | ||||||
Kopecky et al. | 1991 | Kidney TPL | 25
Related posts: Ischemia and Reperfusion Injury in Bone Ischemia and Reperfusion Injury in Bone
 The University of Pennsylvania Classification of Osteonecrosis The University of Pennsylvania Classification of Osteonecrosis
 Alumina Ceramic-on-Highly Cross-Linked Polyethylene Bearing in Cementless Total Hip Arthroplasty in Young Patients with Osteonecrosis of Femoral Head Alumina Ceramic-on-Highly Cross-Linked Polyethylene Bearing in Cementless Total Hip Arthroplasty in Young Patients with Osteonecrosis of Femoral Head
 Impaction Bone Grafting Impaction Bone Grafting
 Causes of Pain in Early and Advanced Stage Osteonecrosis Causes of Pain in Early and Advanced Stage Osteonecrosis
 Kienböck’s Disease of the Lunate Kienböck’s Disease of the Lunate
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||||||