EC Features
Antithrombotic
Prothrombotic
Coagulation protein binding sites
Glycosaminoglycans/ATIII
Binding sites for fibrin, FIX, IXa, X, Xa, FXII, kallikrein
TFPI
Tissue factor
Thrombomodulin
Thrombin receptor
Receptor for protein
C/APC
Products produced and/or stored by platelets
PGI2
vWF
NO
PAF
ADPase
Fibrinogen
FV
FXI
Fibrinolytic factors
t-PA production
PAI-1, PAI-2
u-PA expression
PAI-3 (protein C Inhibitor)
u-PAR
TAFI activation
Plasminogen binding sites
Annexin II
Vasomotor factors
NO
TxA2
PGI2
Endothelin-1
Table 3.2
Extracellular signals that regulate procoagulant or anticoagulant mRNA expression in endothelial cells
Source of signal | Effect |
|---|---|
TNFα | Decreases expression of thrombomodulin |
Increases expression of plasminogen-activator inhibitor type 1 and tissue factor | |
IL-1 | Decreases expression of thrombomodulin |
TGF-β | Decreases expression of thrombomodulin |
Increases expression of plasminogen-activator inhibitor type 1 | |
VEGF | Increases expression of thrombomodulin, plasminogen-activator inhibitor type 1, and tissue-type plasminogen activator |
PDGF | Increases expression of von Willebrand factor |
Shear stress | Increases expression of thrombomodulin, tissue-type plasminogen activator, tissue factor, and nitric oxide synthase |
Hypoxia | Increases expression of plasminogen-activator inhibitor type 1 |
Decreases expression of tissue-type plasminogen activator |
Does the heterogeneity in EC MHCII antigens contribute directly to RED and thrombogenicity? ECs present MHCII antigens to CD4+ T cells—more in micro- than macrovasculature [21, 22]—and some express CD40 (in dendritic cells this marker interacts with T cells), CD80, and CD86 (both T cell costimulatory) suggesting presentation to CD8+ T cells as well [28]. There are conflicting data on the extent to which these interactions anergize the T cells [29, 30]. But there is support for the conclusion that CD8+ T cells can activate and induce proinflammatory changes in ECs [31, 32].
Dendritic cells (DCs) may have a role in multifoci ION. Their role in DIC begins with systemic challenge to the innate immune system. In experiments with lipopolysaccharide (LPS), the challenge begins with detection of the immunogen by DC TLR4. In the lymph nodes LPS activates DC TLR4 that in turn upregulates its protease-activated receptor 1 (PAR1). Through an internal cascade, PAR1 activates sphingosine-phosphate 3 receptor (S1P3R) that initiates secretion of TF and IL-1β. In lymph circulation this TF has little effect. But the continuous LPS signal eventually stimulates migration of DCs from the lymph nodes to lymph vessels that carry them to ducts that drain into the blood circulation. As the DCs circulate they secrete TF stimulating receptive ECs to secrete thrombin and form local thrombi. Thrombin directly stimulates DC PAR1 to complete the autocrine cycle, further amplifying DIC [33].
Local thrombosis may be stimulated by LPS in vessels like the retinacular microcirculation as well. Endothelial cells have TLR4s [34], S1P3R [35], Waeber [36] and PAR1 [37]. This effect has apparently been demonstrated by Okazaki et al. [38] who challenged mice with LPS and were able to produce femoral ION and cytokines associated with TLR4 stimulation.
3.3 Traumatic ION
Thrombogenesis is not necessary for ION if vessels are simply destroyed as is the case for traumatic ION. But bone not directly made ischemic via traumatic destruction of feeder vessels may become ischemic as the result of reperfusion injury . Compartment syndrome would likely produce the necessary ischemia, as would a crush injury of limited force.
Endothelial cells normally control both upstream and downstream vessel diameters by converting L-arginine to NO that is delivered countercurrently or downstream to sphincters. They also normally oxidize their purines using xanthine oxidase to produce superoxide anions, which at physiologic pH dismutates (via superoxide dismutases) to H2O2 and in the presence of ferrous ions and peroxidases to H2O and ferric ions.
During ischemia blood flow in microvasculature stops and neutrophils that have become trapped are no longer stimulated by blood flow shear to remain in the vessel lumen. They upregulate their L-selectin and seek to extravasate through the nearest endothelium. Meanwhile, ECs, in the absence of flow and O2, are increasingly exposed to an anaerobic and decreasing pH environment. As a result NO production and peroxidase activity stops, allowing accumulation of superoxide anion. Accumulation of these free radicals damages ECs, primarily by lipid peroxidation [39].
As tissue adjacent to that made necrotic directly by the trauma proceeds through a wound healing inflammatory phase, constricted vessels upstream begin to relax and blood is shunted to the patent ischemic vasculature. When the highly oxygenated reperfusing blood meets a lumen with neutrophils lining the walls and free radicals and inflammatory cytokines (some from adjacent mast cells) in high concentration, incoming platelets are activated and, in the absence of NO, thrombosis and vessel constriction condemn the flow to stasis.
Selection of vessels that undergo reperfusion injury first is probably not as important for the progress of ION as is the ischemia length of time. We have found significant effects at 2 h [40] as can be seen in Fig. 3.1. These events probably occur at different points and times in the femoral head, depending on local conditions. Diffusion through the extracellular matrix of cytokines and other agents—from mast cells in particular—will spread signals to adjoining tissue. Of particular interest is the source of macrophages, ECs, and fibroblasts that will form the repair tissue of early healing. Unlike idiopathic ION wherein the pathological agent often persists, the trauma source is usually gone when repair begins.
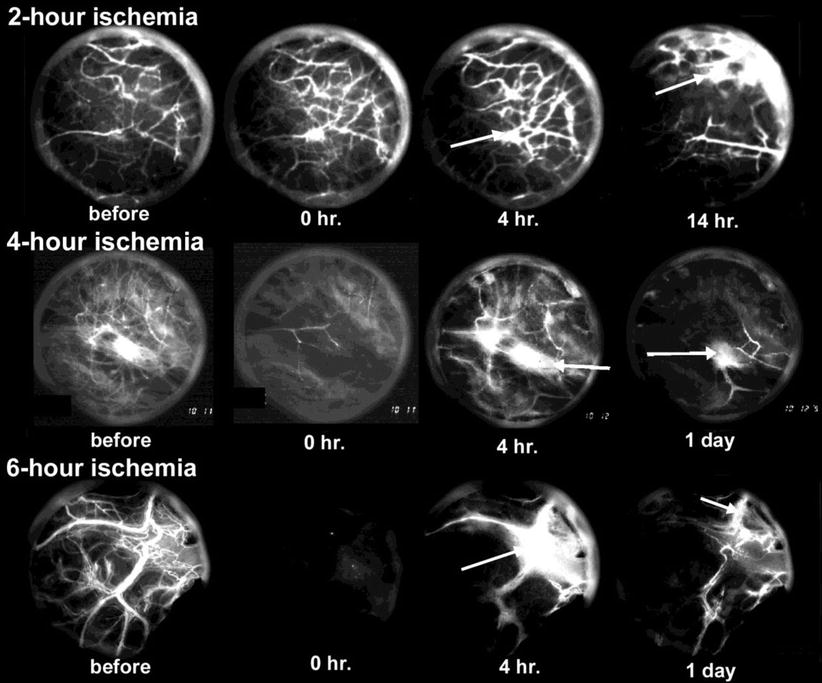
Fig. 3.1
 Distinguishing Bone Marrow Edema Syndrome from Osteonecrosis
Distinguishing Bone Marrow Edema Syndrome from Osteonecrosis
 The University of Pennsylvania Classification of Osteonecrosis
The University of Pennsylvania Classification of Osteonecrosis
 Alumina Ceramic-on-Highly Cross-Linked Polyethylene Bearing in Cementless Total Hip Arthroplasty in Young Patients with Osteonecrosis of Femoral Head
Alumina Ceramic-on-Highly Cross-Linked Polyethylene Bearing in Cementless Total Hip Arthroplasty in Young Patients with Osteonecrosis of Femoral Head
 Impaction Bone Grafting
Impaction Bone Grafting
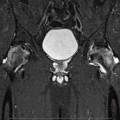 Causes of Pain in Early and Advanced Stage Osteonecrosis
Causes of Pain in Early and Advanced Stage Osteonecrosis
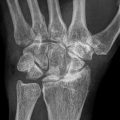 Kienböck’s Disease of the Lunate
Kienböck’s Disease of the Lunate




Examples of reperfusion phase vascularity for three ischemia durations. Each circle has a diameter of about 2 mm (disregarding the outer ring of reflected light). Visible vessels show circulating FITC-D70 which normally leaks only through so-called large pores (according to the two-pore permeability model; few in number in tissue at equilibrium). Baseline vascularity is shown at far left. Reperfusion at zero hours (0 H) is photographed 10 min after cuff deflation and shows a pattern suggesting an inverse relationship between duration and initial reperfusion. At 4 h vessels exhibit abnormal leakage which appears to increase with ischemia duration. At 14 per day, reperfusion has decreased from its 4-h value for all three treatments. Also, leakage at this time is less for the higher durations, probably because so little plasma is circulating (From Hsieh et al. [40] with permission)
Related posts:
Distinguishing Bone Marrow Edema Syndrome from Osteonecrosis
The University of Pennsylvania Classification of Osteonecrosis
Alumina Ceramic-on-Highly Cross-Linked Polyethylene Bearing in Cementless Total Hip Arthroplasty in Young Patients with Osteonecrosis of Femoral Head
Impaction Bone Grafting
Causes of Pain in Early and Advanced Stage Osteonecrosis
Kienböck’s Disease of the Lunate
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
