Fig. 1
Cavities on the β-subunit of HLA-DR10 that were used to develop a series of B cell lymphoma specific SHALs. The three cavities selected for use as ligand-binding sites are shown in yellow, red, and cyan
Many factors are taken into consideration in selecting the sites to ensure that the final SHAL will bind to the target, exhibit the desired selectivity and bind with high affinity. Obviously, optimal binding would be expected if the sites targeted by the ligand components in the SHAL are accessible and are not buried within the folds of the protein. Other factors considered in the selection process include:
(1)
The presence and proximity of two or three neighboring sites, enabling the use of shorter linkers, and thus minimizing the flexibility of the final SHAL.
(2)
The unique size, chemical properties of amino acids surrounding the site (such as positive or negative charged functional groups) and the overall shape of the cavity. This reduces the number of different ligands that might bind into the same cavity and makes the SHAL ultimately more selective.
(3)
The rigidity of the region. Dynamic, highly flexible regions where the peptide chains exhibit a lot of motion may bind many types of ligands and is thus not very selective.
(4)
The depth of the cavity (deeper cavities enable the ligands to interact with more amino acids on the cavity surface thus maximizing the initial affinity of the individual ligand components).
(5)
The hydrophilicity or hydrophobicity of the cavity surface (selecting multiple hydrophobic cavities would lead to the development of SHALs with severe solubility limitations).
The best sites tend to also be epitopes recognized by existing antibodies (as described above for HLA-DR10) or binding sites used by other molecules. Sites targeted by antibodies or bound by other proteins are usually located in the most unique regions of the protein. By selecting these sites, it is also possible to conduct competition experiments (using the antibody or other protein) to test the ligands to ensure they bind to the right region of the protein without having to carry out expensive and time-consuming structural studies. It also provides a mechanism for testing the final SHALs to not only confirm that they bind to the desired site but to also assess their functionality.
3.2 Identification of Ligand Components
Candidate ligands for incorporation into SHALs are identified using a three-step process. The first step is a “virtual” screen in which a commercial database of small molecules such as the Available Chemical Directory (ACD; http://www.symyx.com/products/databases/sourcing/acd/index.jsp) or a public database such as ZINC (http://zinc.docking.org/index.shtml) containing several hundred thousand to millions of small molecule structures are rapidly docked into each cavity and evaluated by the docking program for their fit and potential interactions with the surface atoms surrounding the cavity. The top ranked 1,000 compounds are examined visually to ascertain the predicted interactions are reasonable, and 30–100 of the compounds are selected for experimental testing based on a variety of factors. The particular selection criteria depend on the SHAL’s application. Examples of typical criteria include commercial availability, low cost, the presence of an amino- or carboxyl-group directed away from the protein surface for attachment to a linker, chemical stability, absence of known toxicity, and presence or absence of certain functional groups that might impact the final SHAL’s uptake, biodistribution, or metabolism. These “virtual” screens speed up ligand discovery dramatically and minimize the amount of time and effort required for experimental screening.
The list of compounds selected for each cavity is then tested experimentally to identify, and in some cases rank, those molecules that bind to the purified target protein. A variety of methods have been used to carry out these ligand-binding experiments. Electrospray Mass Spectrometry (EMS) (Loo 1997; Wu et al. 1997; Pramanik et al. 1998; Veenstra 1999; Cosman et al. 2002; Wigger et al. 2002; Shields et al. 2003) and Nuclear Magnetic Resonance (NMR) spectroscopy (Shuker et al. 1996; Roberts 2000; Cosman et al. 2002) require considerable protein, but both methods offer certain advantages over more rapid screening methods such as Surface Plasmon Resonance (SPR) and Dual Polarization Interferometry (DPI). While EMS screening requires that the ligands and protein be ionizable (a significant limitation), the technique is one of the few that provide information about the number of binding sites that can be occupied by one individual ligand. Both EMS and NMR can be used to identify combinations of ligands that are bound simultaneously to the protein. Several different types of NMR experiments have been developed that can be used to confirm ligand binding (Gronenborn and Clore 1982; Meyer et al. 1997; Gounarides et al. 1999; Roberts 2000; Dalvit et al. 2001; Otto and Larive 2001; Cosman et al. 2002), obtain information about the ligand binding site (Medek et al. 2000; Roberts 2000), identify the part of ligand closest to (or farthest away from) the protein surface (Mayer and Meyer 1999; Medek et al. 2000; Roberts 2000) and determine experimentally the distance between bound ligands (Hajduk et al. 1997; Roberts 2000). While both EMS and NMR provide much more information about the bound ligands, the speed and lower cost involved in performing SPR (Schuck 1997; Maynard et al. 2009) and DPI (Swann et al. 2004) screens have contributed to their recent rise in popularity. Both of these methods have been automated and permit the screening of 20–100 ligands per day using μg to sub-μg amounts of protein. Analyses performed with equimolar concentrations of ligands also provide a relative ranking of ligand-binding affinities (Fig. 2).
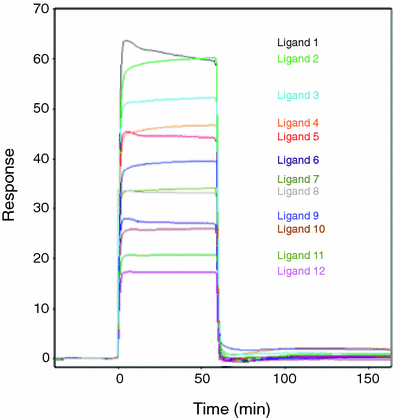
Fig. 2
Ligand binding to the target protein can be tested experimentally using surface Plasmon resonance. In this experiment, the HLA-DR10 protein has been immobilized on the surface of a chip and equimolar concentrations of the 12 ligands (these particular ones were determined in a previous set of experiments to bind to the protein) are pumped into the cell containing the chip one at a time (separated by a rinse buffer to dissociate the previous ligand) and allowed to bind. The height of the binding response (the peak) is proportional to the binding affinity. No response is observed for ligands that fail to bind
Once experimental binding to the protein has been confirmed, each of the ligands is then retested to determine if it can bind to the protein in the presence of an antibody, other protein, or a ligand that normally binds to the targeted site. Such experiments identify those ligands that bind to the right region of the protein. Ligands that do not bind to the protein are not carried forward. The ligands that do bind are then paired into sets and retested to test their ability to bind simultaneously to the target protein in another competition assay. Any of the methods described above can be used to conduct the competition experiments. The resulting ligand pairs that do simultaneously bind to the protein are used to create SHALs.
3.3 SHAL Synthesis and Characterization
The synthetic process that has been developed to link ligands together to create SHALs is a modular solid phase synthetic chemistry approach that employs simple carbodiimide linking reactions to attach one ligand at a time to linkers created from FMOC protected lysine and mini-PEG (Fig. 3) (Balhorn et al. 2007; Hok et al. 2007). Prior to synthesis, the distance separating the pairs or triplets of ligands to be linked together are estimated using the predicted ligand–protein complexes obtained from the docking runs. The separation distance is measured between the functional groups on each ligand bound in their respective cavities. This distance is then used to define the number of lysine and mini-PEG molecules that will be used to interconnect the ligands.
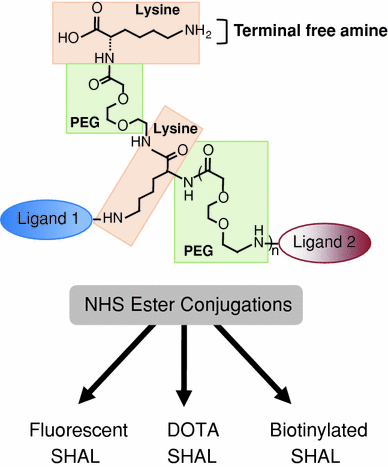
Fig. 3
Modular approach used to synthesize SHALs. The compounds are synthesized using solid phase synthesis. The linkers are built up using a combination of lysine (for branch points) and mini-PEG molecules (to provide length) and the ligands are attached sequentially after deprotecting the appropriate groups at the ends of the linkers. All of the conjugation steps use the same chemistry linking free carboxyl groups to free amino groups. After cleavage of the SHAL from the resin, the resulting terminal free amine can be conjugated to biotin, chelating groups, dyes, or other tags
Lysine is attached to the resin first. In the synthesis of a typical bidentate SHAL, a mini-PEG is attached to this lysine residue’s deprotected free amine followed by the attachment of a second lysine to the end of the mini-PEG. This lysine serves at the branch point on which to construct the “arms” of the linker that separate the two ligands by the required distance. The first ligand is conjugated to the deprotected epsilon amino group on this branch lysine and one or two mini-PEGS, depending on the distance of separation needed, are added to the alpha-amine after deprotection. The second ligand is then attached to the end of the last mini-PEG. Bis-bidentate forms of the SHALs (Fig. 4) can also be synthesized using this same general approach. These are dimeric forms of the molecules that have two SHALs tethered together with a linker sufficiently long to enable the two SHALs to bind to neighboring protein receptors on the cell surface.
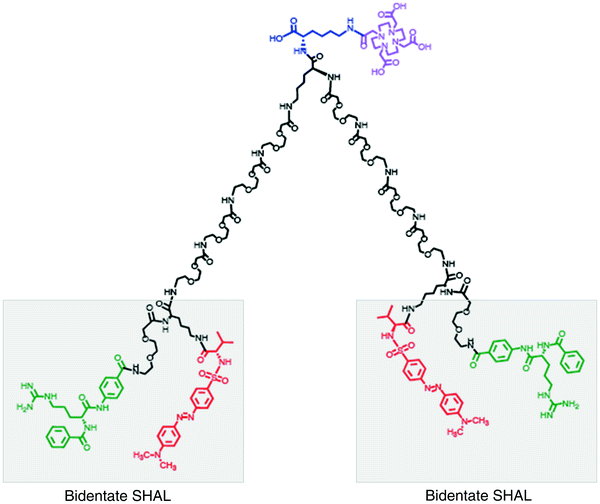
Fig. 4
Dimeric (bis-) forms of the SHALs have been created to mimic the bivalent binding of antibodies to neighboring receptors on the cell’s surface. This bis-bidentate SHAL (DvLPBaPPPP)2LLDo is a dimeric form of DvLPBaPLDo created by linking two bidentate DvLPBaPLA SHALs together with a suitable length linker and attaching the DOTA chelator to the midpoint of the linker
Following cleavage of the completed SHAL from the resin, the sample is purified by high performance liquid chromatography (HPLC). Mass spectrometry is used to identify the HPLC peak containing the SHAL and confirm its mass. The resin in the reaction chamber can be removed and split at any point during the synthesis, making it possible to easily synthesize sets of SHALs with different length linkers (defined by the number of added mini-PEGs) separating the ligands or families of SHALs that share a common ligand. Each SHAL retains a free amino group at the end of the linker. This amine can be readily conjugated to almost any type of tag (e.g., biotin, fluorescent dye, DOTA, or other chelators). Large amounts of SHAL (hundreds of milligrams to grams) can be synthesized rapidly using inexpensive components. New forms of a particular SHAL can also be quickly created by switching out ligands (DeNardo et al. 2007a, b) or attaching other components to lysine branch points inserted into the linker (Balhorn et al. 2009).
3.4 Testing of Prototype SHALs
A combination of protein and cell-based assays has been used to characterize the SHALs designed to target HLA-DR10 (Balhorn et al. 2007; 2009; DeNardo et al. 2007a, b, 2008, 2009; Hok et al. 2007). As a first step, SHAL binding to purified HLA-DR10 protein is confirmed by SPR and the binding affinity is determined. DOTA tagged SHAL is then labeled with a radionuclide (typically 111In) and the binding avidity to tumor cells is determined using cells over-expressing HLA-DR10. Cell selectivity is evaluated by measuring biotinylated SHAL binding to a set of cell lines that express HLA-DR10 and a set that do not express the protein (Fig. 5). The SHALs of interest are then tested for their ability to bind to tumor biopsy sections. Fixed sections are “stained” with a biotinylated form of the SHAL and binding is confirmed using a streptavidin-horse radish peroxidase assay. In contrast to what is observed with Lym-1 and other antibodies that bind HLA-DR10, the SHALs readily bind to formalin-fixed B cell lymphoma tissue without performing antigen retrieval (Fig. 6).
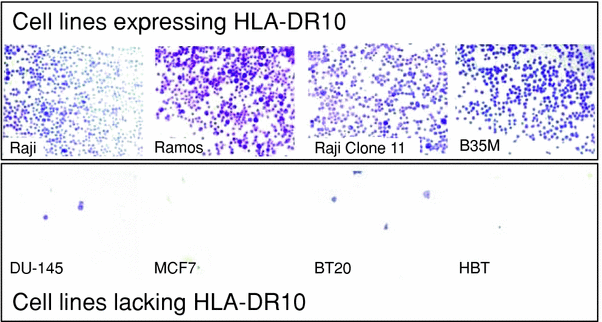
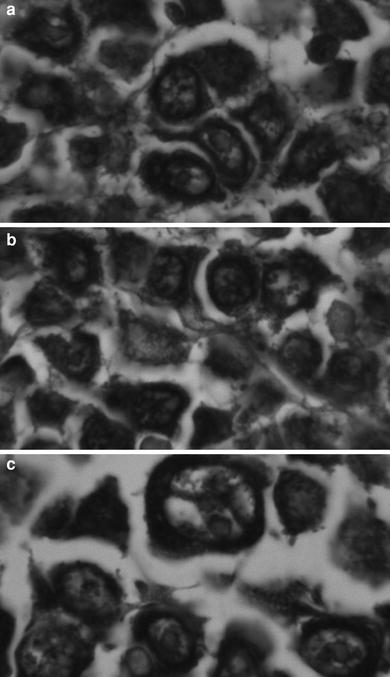
Fig. 5
The selectivity of the SHAL is confirmed using a whole cell binding assay. A biotinylated form of the SHAL is added to ELISA plates coated with streptavidin and the plates are rinsed to remove unbound SHAL. Suspensions of a series of human B cell lymphoma cell lines known to express HLA-DR10 and lack HLA-DR10 are added to different wells on the plates and incubated. The plates are rinsed to remove unbound cells and the bound cells are stained with cresyl violet
Fig. 6
SHAL binding to formalin fixed lymphoma biopsy sections. The tissue sections were treated with the biotinylated form of the tridentate SHAL DvPLCtPCbPLB and the bound SHAL was detected using streptavidin–horseradish peroxidase. The tissue was not counterstained. Bound SHAL is observed as a black precipitate. Cells lacking HLA-DR10 appear unstained (they are not visible). a. Large B cell lymphoma (stomach) from a 55-year-old female. b. Large B cell lymphoma (colon) from a 63-year-old female. c. Large B cleaved cell lymphoma (intestine) from a 70-year-old male
4 SHALs Developed to Date
More than 100 bidentate, tridentate, bis-bidentate, and bis-tridentate SHALs designed to target HLA-DR10 have been synthesized. Our analyses of the results of studies conducted with many of these molecules have begun to provide information about their utility as targeting agents. In every case tested to date, the SHALs designed to target HLA-DR10 have all been shown to be selective, binding to cells expressing HLA-DR10 and not binding to cells lacking this protein (Balhorn et al. 2007, 2009; DeNardo et al. 2007a; Hok et al. 2007). None of the SHALs tested have exhibited toxicity in mice. As shown previously by Fesik and others (Shuker et al. 1996; Kramer and Karpen 1998; Hajduk et al. 1999, 2000a; Fan et al. 2000a, b; Maly et al. 2000; Honma et al. 2001), the linking together of pairs of ligands that bind weakly (with mM to μM affinities) to the protein on their own routinely lead to the production of SHALs exhibiting near nM protein and cell binding affinities. The addition of a third ligand in a tridentate SHAL increased its binding affinity an additional 1,000-fold (Table 1). Similar increases in tumor cell avidity have been achieved through the creation of dimeric (bis) forms of the SHALs. These bis-bidentate SHALs bind to isolated HLA-DR10 protein with affinities similar to the bidentate SHALs, as expected, because the molecules cannot bind bivalently to isolated HLA-DR10 protein. In binding studies conducted with intact Raji cells however, the bis-bidentate SHALs exhibited bivalent binding to neighboring HLA-DR10 proteins on the cell surface with avidities in the pM range.
Table 1
Dissociation constants (Kd) determined for a series of bidentate, bis-bidentate and tridentate SHALs designed to target HLA-DR10
SHAL | Kd (HLA-DR10) | Kd (Raji Cells) |
Bidentate | ||
LePLDB | 50nM | |
DvLPBaLPB | 80nM | |
LeacPLDB | 23nM | |
Bis-bidentate | ||
(DvLPBaPP)2LLDo | 93pM | |
(DvLPBaPPP)2LLDo | 21nM | 51pM |
(DvLPBaPPPP)2LLDo | 18pM | |
(DvLPBaPPP)2LArg6AcLLDo | 34nM | |
Tridentate | ||
DvPLLCtPCbPLB | 23pM |
While only a few of the SHALs have been tested in vivo, several trends have emerged from the pharmacokinetic and tissue biodistribution experiments that have been conducted in mice bearing lymphoma xenografts (DeNardo et al. 2007a, b). As expected for molecules with molecular masses below 4,000 Da, the majority of the bidentate SHALs exhibited blood (t 1/2 ~ 4–7 h), tissue and whole body (t1/2 ~ 6–8 h) clearance times typical of small molecules. Whole-body clearance times for the larger bis-bidentate SHALs were increased two to three fold. The tissue distribution patterns observed for the bidentate and bis-bidentate SHALs appeared to be dictated by the types of ligands used to create the SHALs. SHALs containing peptides accumulated to higher levels in the kidney, while bis-bidentate SHALs containing bile acid-like components accumulated more in the liver. SHALs with a triiodothyronine ligand bound to blood proteins and also accumulated in the liver.
The best characterized and most promising SHAL developed to date is the tridentate molecule DvPLCtPCbPLDo (DeNardo et al. 2008, 2009). The blood clearance rate of this SHAL was found to be similar to that of the bidentate SHALs. In contrast, DvPLCtPCbPLDo exhibited the slowest tumor clearance rate of any SHAL tested. No detectable loss of tumor-bound SHAL was observed over a 24 h period after injection (DeNardo et al., unpublished results). Confocal microscopy studies conducted with cell lines expressing HLA-DR10 (Raji) and lacking HLA-DR10 (Jurkat’s) have shown DvPLCtPCbPLDo is selectively taken up by cells expressing HLA-DR10, and the SHAL accumulates in the cytoplasm. A small amount of the SHAL also appears to enter or become associated with the nucleus. Preliminary observations from an ongoing study evaluating the binding of this tridentate SHAL to several lymphoma tissue arrays indicate the SHAL binds to a large fraction of B cell malignancies. The unlabeled form of this SHAL (with no radionuclide incorporated into the DOTA) has also been shown to be cytotoxic to cell lines (IC50 = 2.5 nM) and to solid lymphoma mouse xenografts expressing HLA-DR10, but not to cells or tumors lacking the protein. Cures have been observed in two-thirds of the mice bearing HLA-DR10-expressing Raji xenografts treated with DvPLCtPCbPLDo that persisted for the lifetime of the mice. DvPLCtPCbPLDo had no effect on tumors lacking HLA-DR10 (Jurkat’s).
In contrast to the majority of other small molecule cancer therapeutics in use today, the cytotoxicity of the radiotherapeutic form of the DvPLCtPCbPLDo SHAL is induced by two different mechanisms. Functioning in a fashion similar to antibodies used for radioimmunotherapy, radionuclides carried by radiolabeled forms of the SHAL destroy the targeted cells by radiation-induced DNA damage. Electron microscopy analyses of xenograft tissue obtained from mice treated with unlabeled tridentate SHAL suggest the second mechanism may involve the same process used by antibodies that target the HLA-DRs, CD20, and a number of other cell surface receptors overexpressed on cancer cells—the induction of a cell signaling event which leads to apoptosis and autophagic cell death. Experiments conducted with a hexa-arginine analog of a bidentate SHAL have demonstrated that additional modifications can be made to SHALs to further enhance their functionality. A hexa-arginine peptide transporter conjugated to the SHAL’s linker increased SHAL uptake by cells. The success of this effort suggests that other molecules, such as small enzyme inhibitors, might also be conjugated to the SHAL linker to create SHAL analogs exhibiting different or additional mechanisms of toxicity.
Related posts:
 (Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
(Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
 Thyroid Carcinoma
Thyroid Carcinoma
 of Tumors: Pretargeting with Bispecific Antibodies
of Tumors: Pretargeting with Bispecific Antibodies
 Imaging for Radiation Therapy Planning
Imaging for Radiation Therapy Planning
 Use of Dosimetry in the Planning of Patient Therapy
Use of Dosimetry in the Planning of Patient Therapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


