Fig. 1
Bispecific pretargeting procedure. After the bsMAb is injected, it will localize in the tumor. In this example, the bsMAb has 2 binding arms for the tumor antigen, with monovalent binding to the hapten. Once the molar concentration of the bsMAb in the blood is low enough, the radiolabeled hapten-peptide is given. The hapten-peptide has 2 haptens that enhance avidity and may even cross-link two adjacent bsMAb (affinity enhancement system, AES). The peptide portion usually contains 4–5 D-amino acids (AA) with a single chelator to capture the radionuclide
An extensive number of preclinical and clinical studies followed this improved method of pretargeting. The bsMAb studied mostly consisted of a murine anti-CEA Fab’ conjugated chemically to the Fab’ of a murine antibody that bound indium-loaded DTPA (Le Doussal et al. 1990). This was used to image a number of different CEA-producing tumors, including colorectal, small-cell lung, and medullary thyroid cancers (MTC) (Le Doussal et al. 1993; Chetanneau et al. 1994; Bardies et al. 1996; Gautherot et al. 1997; Barbet et al. 1998; Gautherot et al. 1998; Hosono et al. 1999; Kraeber-Bodere et al. 1999; Gautherot et al. 2000). Other clinical studies with an 131I-labeled HP provided further evidence that pretargeting could deliver similar radioactivity to tumors compared to directly radiolabeled anti-CEA antibody, but with lower normal tissue accretion (Bardies et al. 1996; Kraeber-Bodere et al.1999; Chatal et al. 1995). Indeed, preclinical studies confirmed this therapeutic advantage (Gautherot et al. 1997, 1998, 2000; Hosono et al. 1999; Kraeber-Bodere et al. 1999; Barbet et al. 1999). Although clinical trials were Phase I studies, a retrospective review of two Phase-I/II pretargeted radioimmunotherapy (PT-RAIT) trials in MTC patients given an 131I-HP found that a subgroup of patients whose calcitonin levels (a sensitive and specific tumor marker for MTC) doubled within two years showed a significant survival advantage over an untreated, comparable, contemporaneous group. This success in a radioresistant cancer was explained by the finding that many of these patients had micrometastatic bone/bone marrow involvement (Mirallie et al. 2005), which was disclosed by pretargeted imaging in 20 of 29 individuals. The overall 10-year survival for patients with bone marrow involvement, as identified by the imaging study at the time of treatment, was significantly longer than those without marrow dissemination of tumor.
These initial studies of bsMAb pretargeting were limited because the bsMAb was of murine origin, and patients developed an anti-mouse antibody response. We then began a collaboration with these French investigators, introducing a Fab’ x Fab’ bsMAb having a humanized anti-CEA coupled to the murine anti-DTPA antibody (Kraeber-Bodere et al. 2006). In contrast to prior studies with the murine bsMAb, where most patients developed an anti-mouse antibody response, only one of 12 patients developed an anti-mouse response, but four developed antibody responses to the humanized anti-CEA portion of the bsMAb.
4 Next Generation bsMAb Pretargeting
We have engineered and examined several different constructs for pretargeting, beginning with bispecific diabodies that had monovalent binding to the target antigen and hapten, then triabodies with divalent binding to the tumor and monovalent binding to a hapten, and now a novel platform technology called the Dock-and-Lock (DNL) method for fusing different proteins recombinantly (Rossi et al. 2003, 2005, 2006) (Fig. 2). For pretargeting, a tri-Fab structure is used, since earlier studies with chemically prepared anti-tumor x anti-hapten bsMAb (e.g., IgG x Fab’ vs. F(ab’)2 x Fab’ and Fab’ x Fab’) suggested that a bsMAb with divalent binding to the tumor is preferred over monovalent binding, but it was also critical that the bsMAb had rapid clearance from the blood (Karacay et al. 2002). Divalency of the HP also provided enhanced avidity.
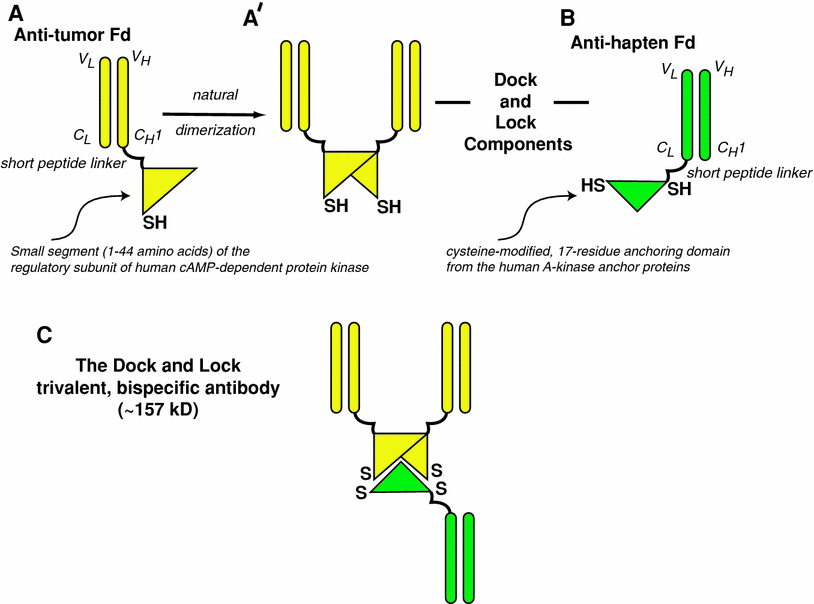
Fig. 2
Bispecific antibodies based on the Dock-and-Lock procedure. The Fd portion of an anti-tumor antibody (Fd is the full light chain associated with the VH and CH1 portion of the heavy chain) is linked to a short peptide that tethers the dimerization-docking-domain of the regulatory subunit of the human cAMP-dependent protein kinase A (a). This amino acid sequence was modified to insert a cysteine residue in a strategic location. The recombinant anti-tumor Fd naturally forms non-covalent dimers (b), with the resulting complex providing a “docking” site that binds to a 17-amino acid sequence derived from the A-kinase anchor proteins. This sequence, which is specially modified to place two cysteine residues in strategic locations, is tethered to the Fd portion of the anti-hapten antibody (c). When the anti-tumor and anti-hapten proteins are mixed, a bispecific tri-Fab (TF) 157-kD antibody is formed, with a specific orientation that places the cysteine residues in position that allows for the formation of covalent disulfide bonds
BsMAb pretargeting has relied on the ability of the bsMAb to clear from the blood naturally, usually in 1–2 days in mice for a 100-kD Fab’ x Fab’ bsMAb, and 4–5 days in humans (Kraeber-Bodere et al. 2006; Karacay et al. 2000). Although such tri-Fab bsMAbs have the same molecular weight as IgG (~157 kD), they also were found to clear very rapidly from the blood, with <1% per gram of the injected dose present in the circulation within 1 day (Rossi et al. 2006; Gold et al. 2008; Sharkey et al. 2008a, b, 2009). These tri-Fab bsMAbs were stable in serum in vitro and in vivo, as determined by high-pressure liquid chromatography. We hypothesize, therefore, that the rapid clearance is at least partially due to the absence of an Fc in the tri-Fab structure.
Due to this rapid clearance, tumor accretion is much lower than for IgG, reaching a maximum within 6 h (Gold et al. 2008). The protein dose can be increased to load the tumor with more bsMAb to maximize the amount of radiolabeled HP captured. This has been defined as the bsMAb/peptide mole ratio, which expresses the moles of bsMAb needed with a prescribed amount of radiolabeled HP (Sharkey et al. 2003a, b). In our experience, most tumor xenograft systems achieve the highest tumor accretion when 10- to 20-fold more moles of bsMAb than HP moles are given. Experimentally, between 1 and 4% of the bsMAb is bound per gram tumor when the radiolabeled HP is injected, yet 10–30% per gram of the HP can be captured in the tumor. On a mole basis, this calculates to a mean of 1 mol of HP per 1–2 mol of bsMAb. Since the bsMAb has monovalent binding to the hapten and the HP is divalent, each bsMAb in the tumor is occupied with the HP, under optimal conditions.
Dynamic imaging indicated that the HP penetrates the tumor within 5 min, having maximum uptake within 30–60 min, and at least 60% excreted in the urine at 1 h (Sharkey et al. 2005a, b). In contrast, a radiolabeled Fab’ shows very little tumor accretion within an hour. Thus, with the radiolabeled HP traversing the vascular space efficiently, and given sufficient bsMAb for capturing the small amount of HP that passes the tumor, pretargeting can provide a large therapeutic window within only an hour that can be sustained over several days.
Timing of the radiolabeled HP injection is another factor determining optimization of the procedure. The HP binds to the anti-hapten binding arm of the bsMAb, so an excess of bsMAb in the blood would result in preferential binding of the HP to the circulating bsMAb. Thus, with bsMAb pretargeting, there is a level where the radiolabeled HP can interact with the bsMAb in the blood. Preclinically, we found that the highest tumor accretion occurred when administration of the radiolabeled HP is delayed until the bsMAb has cleared to a concentration of <1% per gram in the blood (Karacay et al. 2000; Sharkey et al. 2003a, b). When this is exceeded, tumor accretion decreases with higher blood concentrations. If the delay is too long, then there is a risk that there will be a decrease in concentration of the bsMAb in the tumor, so that the level for capturing the radiolabeled HP will not be optimal. However, even with less optimal pretargeting conditions, tumor/nontumor ratios are often much better than with a directly radiolabeled antibody.
In order to predict how the bsMAb and HP interact, it is better to use their molar concentrations rather than a percentage. In this case, in the HSG (histamine-succinyl-glycine)-hapten system we have examined, we found that when the molar concentration of the bsMAb in the blood is at least 10-fold lower than the estimated molar concentration the HP will be at the instant it is injected (e.g., nmoles of peptide divided by estimated total blood volume), the HP will clear at a rate as if no bsMAb were present. This clearance is exceptionally fast (minutes). As the bsMAb concentration increases, the HP’s clearance will gradually become slower, but still, more than 90% of the HP can be cleared in less than 1 day when the bsMAb concentration is 10-fold higher than the HP. This interaction is governed by the affinity of the hapten-anti-hapten antibody. However, it is important to note that at the usual 10−9 M affinity constant for most antibodies, the HP-bsMAb is reversible, and thus the HP does not bind fastidiously to the bsMAb in the blood. This is in sharp contrast to the avidin–biotin bond that is used in other pretargeting systems, where with a very high affinity of 10−15 M, complexes formed in the circulation would not likely to be uncoupled. Hence, a clearing agent is used in systems that employ avidin–biotin. Ensuring the radiolabeled HP clears rapidly from the blood is an essential requirement for optimizing pretargeting systems, since minimizing the residence time in the red marrow will mitigate hematological toxicity. Therefore, we strive to administer the radiolabeled HP when the bsMAb concentration in the blood is minimal.
Another key factor of pretargeting is the need to maximize the HP’s specific activity (e.g., Ci/mmole). The specific activity directly affects the radioactivity targeted with the HP; the lower the specific activity, less radioactivity is delivered per unit of HP. As already discussed, the optimum moles of bsMAb given depend on the moles of HP administered, and therefore the bsMAb dose would have to be increased to capture the same amount of radioactivity if the HP were prepared at a lower specific activity. We already have seen how this can negatively impact tumor uptake and therapy in mouse xenograft model system using a 177Lu-labeled HP (Schoffelen et al. 2010). In this model, the therapeutic dosing was started with a single injection of the bsMAb followed by the HP. As the 177Lu-HP was increased, the animals showed no evidence of toxicity, but escalation could not continue further because in order to maintain an optimal bsMAb/HP mole ratio, the bsMAb would need to be increased to a level higher than that required to saturate all antigen in the tumor, and thus the amount of 177Lu-HP localized in the tumor would not increase commensurately. The solution to this dilemma was to have a delay before giving another bsMAb/177Lu-HP dosage. Using this fractionated approach improved therapeutic responses.
The nature of the hapten used is also very critical in pretargeting. We have focused on the use of HSG as the hapten, using the anti-HSG antibody, 679, as part of the bsMAb. Earlier hapten systems used antibodies to specific chelate-metal complexes, but because these systems were so specific for the chelate-metal complex, a different anti-hapten antibody would have been required if another radiometal were to be used. HSG is not involved in binding of the radionuclide. Thus, a HP containing HSG can have any number of radionuclide-binding ligands for capturing a variety of different radionuclides for imaging or therapy. The anti-HSG 679 antibody has an acceptable affinity to HSG (e.g., ~10−9 M), but a very weak affinity to histamine (Morel et al. 1990; Janevik-Ivanovska and Gautherot 1997). Initially used as part of a radioiodinated derivate for pretargeting (Hosono et al. 1999; Janevik-Ivanovska and Gautherot 1997), a number of other HSG-peptides have been introduced to capture a variety of other radionuclides, including 99mTc, 111In, 90Y, 177Lu, 124I, 68Ga, and 18F (Sharkey et al. 2003a, b, 2005a, b; Gestin et al. 2001; Griffiths et al. 2004; Morandeau et al. 2005; McBride et al. 2006, 2009, 2010; Schoffelen et al. 2010; Karacay et al. 2011). Preclinical studies indicated the anti-HSG antibody did not bind to human tissues, and peptides containing HSG did not have histamine-like pharmacological activity (Sharkey et al. 2010).
During the period required to clear the bsMAb from the blood, the bsMAb must remain accessible (i.e., on the surface or within the interstitial space of the tumor) in order to bind the HP. While most bsMAb pretargeting procedures have used an interval of several days for the bsMAb to clear from the blood and tissues, other pretargeting methods have a clearing step to remove the pretargeted agent from the blood after it localizes in the tumor (Sharkey et al. 2005a, b). Studies have found bsMAb pretargeting could also benefit from a clearing step (Mirallie et al. 2005; Goldenberg et al. 2006), but we have avoided this because of the added complexity. Pretargeting methods utilizing streptavidin/avidin and biotin incorporate a clearing step, because the very high affinity between these agents results in very stable complexes in the blood that may affect tumor targeting.
If the binding of a bsMAb to the antigen is not retained on the surface of the tumor, but is internalized, HP uptake could be compromised. While internalizing targets might not be ideal for pretargeting, acceptable pretargeting results have been achieved using an antibody against EGP-1 (also known as Trop-2 or GA733-1), RS7, that readily internalizes (Govindan et al. 2004; Stein et al. 1999). A tri-Fab bsMAb targeting EGP-1 and HSG, TF12, successfully localized a radiolabeled HP to human ovarian and prostate cancer cell lines (Sharkey et al. in press). However, other internalizing antibodies, such as anti-CD22, have not been found to give satisfactory pretargeting when examined in three different human lymphoma cell lines against antibodies against other B cell antigens (Pagel et al. 2009). In this case, it is uncertain whether internalization of CD22 was the complete explanation for this difference, since the affinity of the anti-CD22 antibody was nearly 10-fold less than that of the anti-CD20 antibody, and CD22 density on these cell lines was also much lower than CD20 (Pagel et al. 2009; Pantelias et al. 2007). Nevertheless, while targeting methods likely benefit from more abundant and accessible target antigens, these findings suggest that each antibody/antigen model needs to be evaluated to make this determination.
It is also important to appreciate the role played by tumor and vascular physiology when defining the best conditions for tumor localization. Tumor vascular permeability is particularly important for most antibodies and other targeting agents. In one model studied, vascular permeability and blood vessel density proved to be more important for pretargeting uptake of the HP than antigen density on the tumor cells (van Schaijk et al. 2005). Despite the high uptake of the HP within 1–4 h, a rapid washout from the tumor, reducing the percent injected dose per gram from 300 to 100% in 24 h, was recorded. Thus, such targeting systems have a complexity that defy simple prediction as to targeting success based only on antigen density, degree of antibody internalization, etc. (Sharkey 2005).
We have examined at least five different tumor models/antibodies with bsMAb pretargeting, such as directed against CEACAM5, or CEA (Rossi et al. 2003, 2005, 2006; Sharkey et al. 2003a, b, 2005a, b, 2008a, b; McBride et al. 2006, 2009; Karacay et al. 2005), CSAp (colon-specific antigen-p) (Sharkey et al. 2003a, b), CD20 (Sharkey et al. 2005a, b, 2008a, b, 2009), a pancreatic mucin (antibody PAM4) (Gold et al. 2008; Karacay et al. 2009), and EGP-1 (or Trop-2) (Sharkey et al. in press). In non-Hodgkin lymphoma, as well as colorectal and pancreatic cancers, exceptional localization resulting in improved imaging and therapy has been observed. The therapeutic advantage is due primarily to two attributes. First, the area under the curve (AUC) for the red marrow is very low, in contrast to directly radiolabeled IgG, permitting higher doses of 90Y to be administered (e.g., 0.7 to 0.9 mCi for HP pretargeted vs. 0.15 mCi for a directly radiolabeled antibody) (Sharkey et al. 2008a, b; Karacay et al. 2005, 2009). The AUC for the tumor is often 1.3 to 1.5-fold higher in the pretargeted animals at the maximum tolerated dose given, compared to the directly radiolabeled IgG. However, another advantage is that radiation is deposited in the tumor rapidly, with maximum accretion within 1 h, which means that the radiation is immediately localized, providing the tumor with a 2- to 3-fold higher dose rate than when a directly radiolabeled antibody is given (Sharkey et al. 2005a, b). Although pretargeting delivers the radiation rapidly, the rate of dissociation is usually faster than that of a whole IgG, which can require 1–3 days to reach maximum tumor levels. In some models, the radioactivity delivered by a directly radiolabeled antibody can remain at a high level for several days. Careful matching of the physical half-life of the radionuclide with the residence time in the tumor, as with any radionuclide-targeting system, provides the most optimal therapy. Weekly fractionation of the pretargeted radiolabeled HP can improve responses as compared to single treatments (Schoffelen et al. 2010; Karacay et al. 2009).
5 Other Pretargeting Systems
While the pretargeting concept started with bsMAb, there are now other methods of pretargeting, such as with avidin or biotin bound to targeting antibodies and then the radionuclide-conjugated biotin or avidin, respectively, being administered. This approach, which suffers from the immunogenicity of avidin (or streptavidin), nevertheless has shown good imaging and therapy results in single-cycle use, but has required a “clearing” agent after injecting of the targeting antibody (Goldenberg et al. 2006). Still another method of pretargeting has involved oligomer/complimentary pretargeting, but has been studied only in preclinical models (He et al. 2010; Liu et al. 2010a, b). A summary of the principal methods of pretargeting that have advanced to clinical trials for cancer detection and therapy has appeared elsewhere (Goldenberg et al. 2006), so will not be discussed further in this chapter.
6 Concluding Remarks
Radiolabeled antibody targeting was one of the first selective therapies developed for cancer, beginning with a one-step use of a directly radiolabeled antibody. However, there is expanding preclinical and now clinical evidence that pretargeting procedures can offer distinct advantages by being more effective therapeutically without increased toxicity, as well as able to improve the signal/noise ratio for improved imaging. Thus, we believe that the additional effort involved in a 2-step procedure, separating the targeting bsMAb from the delivery of the therapeutic hapten-peptide, is justified by the outcome observed so far. Whether these procedures will ultimately become a part of the cancer therapeutic armamentarium will depend, of course, on further clinical experience. Animal models already indicate very promising results for pretargeted therapy of non-Hodgkin lymphoma (Sharkey et al. 2008, 2009; Pagel et al. 2008; Press et al. 2001), a tumor where directly radiolabeled antibodies have been successful clinically. Since recent clinical trials with directly radiolabeled antibodies have shown that they can be given in combination with chemotherapeutic regimens that are limited by hematological toxicity (Perrotti et al. 2009; Winter et al. 2009; Jacobs et al. 2008; Morschhauser et al. 2008; Peyrade et al. 2008; Zinzani et al. 2008a, b; Krishnan et al. 2008), the reduced red marrow doses resulting with pretargeting methods could provide an advantage in this setting. Indeed, improved therapeutic responses have been achieved in animal models of solid tumors when pretargeted radioimmunotherapy was combined with chemotherapy (Karacay et al. 2009; Graves et al. 2003; Kraeber-Bodere et al. 2002). Hence, pretargeted radioimmunotherapy is a promising new technology for improved radionuclide therapy in oncology, but may also have a potential for improved drug targeting.
Related posts:
 (Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
(Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
 Thyroid Carcinoma
Thyroid Carcinoma
 of Progressive Dedifferentiated and Medullary Thyroid Cancer with Radiolabeled Somatostatin Analogs
of Progressive Dedifferentiated and Medullary Thyroid Cancer with Radiolabeled Somatostatin Analogs
 Bisphosphonates for Bone Pain Palliation
Bisphosphonates for Bone Pain Palliation
 Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
Radiopharmaceuticals for PET Dosimetry Before, During, and After Endoradiotherapy
 Use of Dosimetry in the Planning of Patient Therapy
Use of Dosimetry in the Planning of Patient Therapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


