Therapeutic nuclide
Diagnostic nuclide (positron emitter)
Nuclide
Half-life
Nuclide
Half-life
β+ branching (%)
Availability
Isotopic substituent
131I
8.04 d
124I
4.18 d
23
Cyclotron
122I
3.62 min
77
Cyclotron, generator 122Xe/122I
67Cu
2.58 d
62Cu
9.74 min
97
Cyclotron, generator 62Zn/62Cu
90Y
2.67 d
86Y
14.74 h
33
Cyclotron
47Sc
3.34 d
44Sc
3.93 h
94
Generator 44Ti/44Sc
Non-isotopic substituent
90Y
2.67 d
153Sm
1.95 d
68Ga
67.7 min
89
Generator 68Ge/68Ga
177Lu
6.71 d
44Sc
3.93 h
94
Generator 44Ti/44Sc
213Bi
45.6 min
110mIn
1.15 h
62
Generator 110mSn/110mIn
225Ac
10.0 d
3 Overview of Generator-Produced Positron Emitters
A radionuclide generator is a concept defined as an effective radiochemical separation of decaying parent and daughter radionuclides such that the daughter is obtained in a pure radionuclidic and radiochemical form. Relevant developments have been reviewed by Rosch and Knapp (2003). Among the generator pairs relevant for quantitative PET, all daughter nuclides are positron emitters and provide significant positron branching, although in some cases the decay is accompanied by high-energy photons, which might require careful adoption of PET scanners. Parent nuclides are neutron deficient and are thus typically produced at accelerators.
Table 2 categorizes the most relevant “PET generators” according to the half-life of the daughter nuclide. As the short half-lives in the range of minutes do not allow radiochemical synthesis, these systems are relevant for perfusion imaging exclusively. In contrast, the longer lived daughter may be used to synthesize labeled compounds which are chemically or physiologically analogous to the radiotherapeutic compounds used today for patient treatment. The longer lived daughter nuclides, on the other hand, provide a potential for the development of labeled radiopharmaceuticals matching the physical half-life of the therapeutic radionuclides.
Table 2
Generator-produced positron emitters with potential for PET imaging
Generator system | Parent | Daughter | |||
|---|---|---|---|---|---|
T ½ | T ½ | 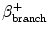 (%) (%) | E β+ (MeV) | Application | |
82Sr/82Rb | 25.6 d | 1.27 min | 95.0 | 1.41 | Perfusion |
140Nd/140Pr | 3.37 d | 3.39 min | 51.0 | 0.544 | Perfusion |
118Te/118Sb | 6.00 d | 3.6 min | 74.0 | 0.882 | Perfusion |
122Xe/122I | 20.1 h | 3.6 min | 77.0 | 1.09 | (Labeling) |
128Ba/128Cs | 2.43 d | 3.62 min | 69.0 | 0.869 | Perfusion |
134Ce/134La | 3.16 d | 6.4 min | 63.0 | 0.756 | Perfusion |
62Zn/62Cu | 9.26 h | 9.74 min | 97.0 | 1.28 | Labeling; perfusion |
52Fe/52mMn | 8.28 d | 21.1 min | 97.0 | 1.13 | Perfusion |
68Ge/68Ga | 270.8 d | 1.135 h | 89.0 | 0.74 | Labeling; perfusion |
110Sn/110mIn | 4.1 h | 1.15 h | 62.0 | 0.623 | Labeling |
44Ti/44Sc | 60 ± 3 a | 3.927 h | 94.0 | 0.597 | Labeling |
72Se/72As | 8.4 d | 1.083 d | 88.0 | 1.02 | Labeling |
Some generator pairs have been proposed decades ago and have continuously been improved radiochemically. Today, a few generators are in routine clinical use, such as 62Zn/62Cu, 68Ge/68Ga, and 82Sr/82Rb. Several others have been studied intensively concerning radiochemical parameters. Improved versions are described, for example, for 44Ti/44Sc, 52Fe/52Mn, 72Se/72As, 140Nd/140Pr. In the context of quantitatively monitoring radiotherapeutic procedures, however, only a few of them are relevant because of specific reasons:
Isotopic matching Ideally, the positron emitting generator daughter should be another isotope of the therapeutic isotope. This is the case for e.g., 122I versus 131I, and 62Cu versus 67Cu. However, there is a significant discrepancy between the half-lives of the corresponding diagnostic and therapeutic isotope, i.e., 3.6 min (122I) and 8.04 d (131I) or 9.74 min (62Cu) versus 2.58 d (67Cu), respectively.
Me(III) analog chemistry These are the ones generating trivalent radiometals, namely 68Ga and 44Sc and eventually 110mIn, to simulate radiotherapeutics based on trivalent particle emitters such as 90Y, 153Sm, 177Lu (and other trivalent lanthanide radionuclides), 213Bi, 225Ac (and other trivalent actinide radionuclides). The therapeutic β− or α-emitting radionuclides are usually attached to molecular tumor targeting vectors such as peptides or proteins (mab, mab fragments) via bifunctional chelators like DTPA or macrocyclic versions such as DOTA. The concept in this case is to consider the coordination chemistry of the “original” trivalent therapeutic radionuclides comparable to the coordination chemistry of the positron emitter. Whether the resulting biological parameters, such as in vitro binding affinities of the Me(III) analog compounds, are still reflecting sufficiently the parameters of the original therapeutics needs to be studied in each case. However, if there is a sufficient homology, the physical half-lives of 67.7 min, 1.15 h, and 3.97 h, respectively, of 68Ga, 110mIn, and 44Sc, may guarantee excellent PET imaging within a few hours p.i. (68Ga) or almost 1 day (44Sc).
In vivo generators This concept involves labeling of molecular carriers (complexes, peptides, mcab, mcab-fragments, etc.) with an intermediate half-life generator parents nuclide, which after accumulation in the desired tissue generates the much shorter half-life daughter radionuclides. It may combine the longer physical (and biological) half-life of the parent nuclide with a short half-life of the positron emitting daughter nuclide.
4 Generators for Isotopic Matching
122 Xe (T ½ = 20.1 h) / 122 I (T ½ = 3.6 min). Various positron emitting nuclides of iodine have been proposed to obtain precise data on localization, quantification, and dosimetry using PET. 122I represents an important example providing a 77 % β+ branching. The availability via the generator was investigated as a by-product during the production of the 123Xe / 123I generator by the 127I(p,6n)122Xe reaction with 70 MeV protons (Diksic and Yaffe 1977; Richards and Ku 1979; Mathis et al. 1986; Lagunas-Solar et al. 1986) or analogously via the (d,7n) channel (Weinreich et al. 1974) and the 124Xe (p,3n) process (Tarkanyi et al. 1991). Amphetamine analogs and iodoperidol were successfully labeled with 122I to measure brain blood flow with PET (Braun et al. 1977; Mathis et al. 1985; Moerlein et al. 1987). Not only rapid iodination chemistry is required to make use of the short-lived iodine positron emitter, but also the short physical half-life might not correspond to longer lasting physiological requirements. Most problematic, however, is the availability of such a generator system to work in a clinical environment.
62 Zn (T ½ = 9.26 h) / 62 Cu (T ½ = 9.74 min). The parent 62Zn is produced via the (p,2n) reaction on 63Cu (Robinson et al. 1980) or via the 60Ni(α,2n) process (Neirinckx 1977; Zweit et al. 1992). Anion exchange chromatography is clearly the method of choice for this generator system. To adsorb no-carrier-added 62Zn2+, Dowex 1 was used and Cu2+ is eluted with hydrochloric acid (Yagi and Kondo 1979; Rhamamoorthy and Mani 1981; Fujibayashi et al. 1989; Green et al. 1990). CG-120 Amberlite resin provided elution of 62Cu in 70 % yield with a glycine solution (Fujibayashi et al. 1989) allowing subsequent ligand exchange reactions. For elution, other ligands (Okazawa et al. 1994) or HCl/ethanol solutions (Zweit et al. 1992) were also introduced. A recent publication (Fukumura et al. 2006), described further refinement of this generator, and focused on high activity (1.8–3.5 GBq) loading of the 62Zn2+ on a Sep–Pak™ “plus” CM weak acidic cation exchanger column with a high 62Cu elution yield of ~96 % with a low 3 mL volume of 200 mM glycine solution. The Zn parent breakthrough is <0.1 %. Semi-automated syringe pump systems were designed for separated hot cell separation system and dispensing systems.
The 62Cu was chelated to human serum albumin (HSA) (Fujibayashi et al. 1990) and benzyl-TETA-HSA (Mathias et al. 1991) and used for blood pool imaging (Herrero et al. 1996). Several hypoxia and perfusion 62Cu-complexes have been developed and applied for PET studies such as [62Cu]ATSM (diacetyl-bis(N4-methylthiosemicarbazone)) (Fujibayashi et al. 1997) and [62Cu]PTSM (pyruvaldehyde bis(N4-methylthiosemicarbazone)) (see, e.g., Green et al. 1990; Mathias et al. 1990; Shelton et al. 1989, 1990; Bormans et al. 1992; Taniuchi et al. 1997). Human biodistribution and dosimetry studies of the perfusion agent [62Cu]PTSM were investigated and this tracer was recommended for repeated studies of myocardial imaging in the same patient (Wallhaus et al. 1998; Wallhaus et al. 2001; Haynes et al. 2000) as well as for quantification of the cerebral blood flow (Okazawa et al. 1994). The compound was also applied for the assessment of angiotensin II-induced blood flow changes in patients with colorectal liver metastases (Flower et al. 2001).
However, until now there was no intention to use the generator-derived 62Cu as a PET-surrogate to therapeutic 67Cu.
5 Generators for Me(III) Analog Chemistry
110 Sn (T ½ = 4.1 h) / 110m In (T ½ = 1.15 h). Positron emitting 110mIn could be a choice for quantitative imaging (Tsai et al. 1997; Lubberink et al. 2002a, b). The direct production of 110mIn via 110Cd(p,n)-, 107Ag(α,n)- ,and 109Ag(3He,2n)-processes, however, leads to the co-formation of the ground state isomer 110gIn (T ½ = 4.9 h). Isotopically pure 110mIn could only be prepared via the generator system 110Sn/110mIn (Tsai et al. 1997). 110Sn was produced via the 110Cd (3He, 3n) reaction with 36 → 25 MeV 3He particles and 110SnCl4 was isolated thermo-chromatographically from the enriched target material. The generator itself was designed by adsorption of 110Sn on a small Kieselgel 40 column and 110mIn was eluted quantitatively within 1 mL 0.02 M HCl.
68 Ge (T ½ = 270.8 d) / 68 Ga (T ½ = 68 min). This generator system has found widespread application, mainly for the synthesis of DOTA- or NOTA-conjugated octreotide derivatives. A first compilation of the data relevant to 68Ge / 68Ga generator systems was published in 1996 (Mirzadeh and Lambrecht 1996). The IAEA has recently initiated comprehensive review of the production of several generator mother nuclides including a chapter on 68Ge (Rösch and Filosofov 2010).
Integral 68Ge thick targets yields have been calculated (Horiguchi et al. 1982) for accelerator-based nuclear reactions on natural metallic germanium and zinc, and Ga2O3 targets. The thick target yields for the Ga(p,2n) reactions have been recently validated (IAEA 2001). Experimental thick target yields—calculated for a 1 h irradiation as well as for saturation—amount to 0.96/0.53 MBq μA−1 h−1 at this energy, but reach values of >2/> 1 MBq μA−1 h−1 already at 25 MeV, respectively. Due to the long half-life of 68Ge, high current accelerators are required for sufficiency of the parent nuclide. Routine production is established at Brookhaven National Laboratory (BNL) and Los Alamos National Laboratory (LANL), USA, iThemba Laboratories/NAC, Faure, Republic of South Africa, and Obninsk, Russian Federation. Those production centers report on production capacities of about 18.5–74 GBq (0.5–2 Ci) 68Ge per batch. At the Cyclotron Co., Ltd., Obninsk, Russian Federation, gallium-nickel alloys are used as target material, prepared on copper backings. Irradiations are performed at rather high proton beam intensity of several hundred μA at 23 MeV proton energy. 68Ge of high specific activity of >74 GBq (>2 Ci)/mg and 99.8 % radionuclidic purity is obtained (Razbash et al. 2005).
Related posts:
 (Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
(Yttrium-90 Microspheres) for Metastatic Hepatic Malignancies
 Thyroid Carcinoma
Thyroid Carcinoma
 of Tumors: Pretargeting with Bispecific Antibodies
of Tumors: Pretargeting with Bispecific Antibodies
 Imaging for Radiation Therapy Planning
Imaging for Radiation Therapy Planning
 Use of Dosimetry in the Planning of Patient Therapy
Use of Dosimetry in the Planning of Patient Therapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


