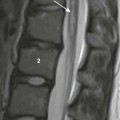
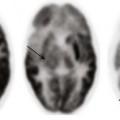
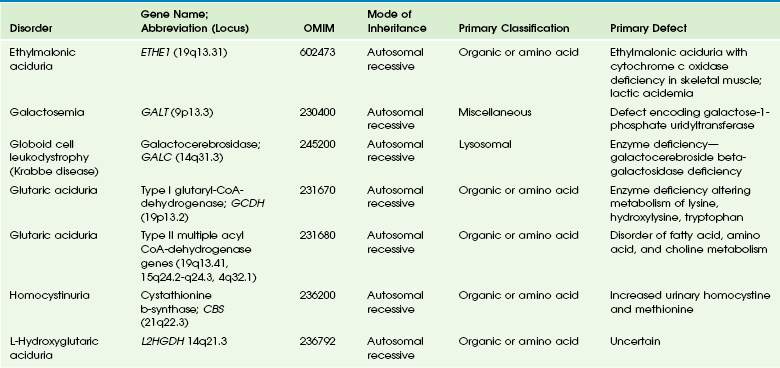
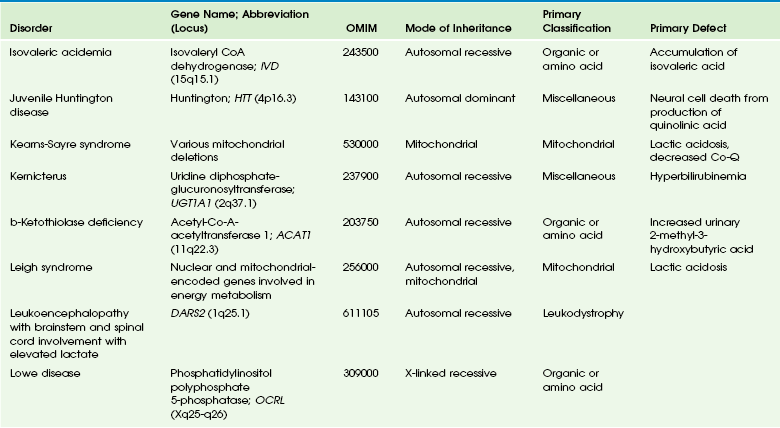
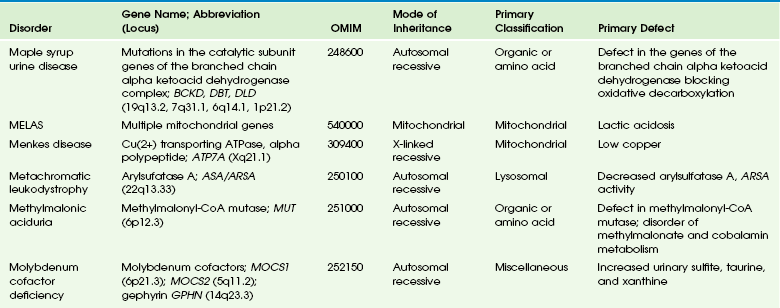
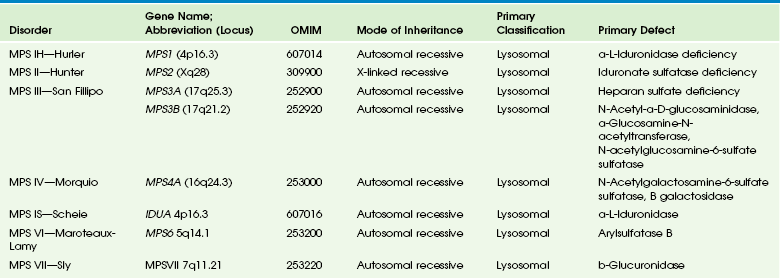
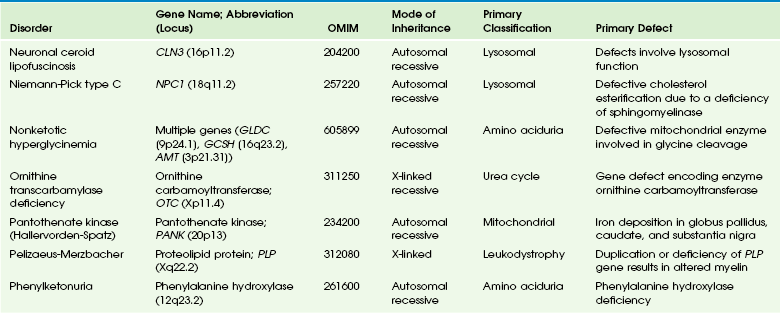
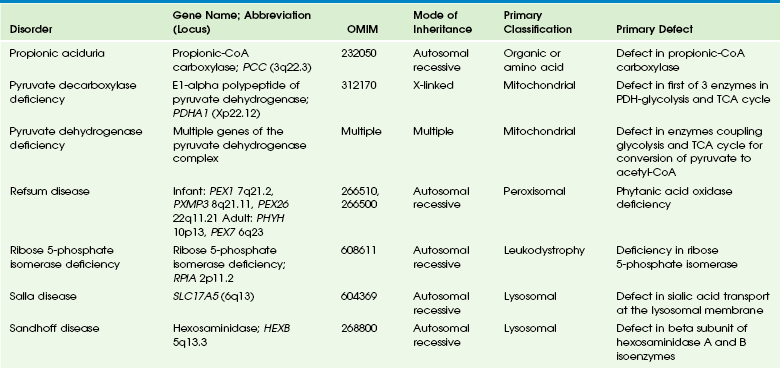
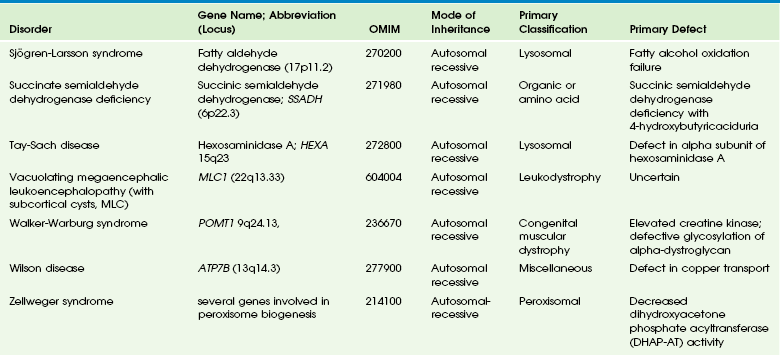
Chapter 33 This chapter includes as a reference a large listing of metabolic brain diseases encountered in the imaging setting (see Tables 33-1 to 33-10). For convenience, the recognized metabolic or genetic defect and the patterns of inheritance are summarized. However, because of space limitations, the following discussion will address only the most commonly encountered of these rare disorders. Table 33-1 OMIM, Online Mendelian Inheritance in Man Database; VLCFA, very-long-chain fatty acid. Adapted From Cecil KM. MR spectroscopy of metabolic disorders. Neuroimaging Clin North Am 2006;16:87-116; used with permission. Data from The Johns Hopkins University, Online Mendelian Inheritance in Man (OMIM), McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and the National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. Available at http://www.ncbi.nlm.nih.gov/omim/. Accessed October 2012. Table 33-2 Adapted From Cecil KM. MR spectroscopy of metabolic disorders. Neuroimaging Clin N Am 2006;16:87-116; used with permission. Data from The Johns Hopkins University, Online Mendelian Inheritance in Man (OMIM), McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and the National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. Available at http://www.ncbi.nlm.nih.gov/omim/. Accessed October 2012. Table 33-3 DNA, Deoxyribonucleic acid; OMIM, online Mendelian Inheritance in Man Database. Adapted From Cecil KM. MR spectroscopy of metabolic disorders. Neuroimaging Clin N Am 2006;16:87-116; used with permission. Data from The Johns Hopkins University, Online Mendelian Inheritance in Man (OMIM), McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and the National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. Available at http://www.ncbi.nlm.nih.gov/omim/. Accessed October 2012. Table 33-4 OMIM, Online Mendelian Inheritance in Man Database. Adapted From Cecil KM. MR spectroscopy of metabolic disorders. Neuroimaging Clin N Am 2006;16:87-116; used with permission. Data from The Johns Hopkins University, Online Mendelian Inheritance in Man (OMIM), McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and the National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. Available at http://www.ncbi.nlm.nih.gov/omim/. Accessed October 2012. Table 33-5 OMIM, Online Mendelian Inheritance in Man Database. Adapted From Cecil KM. MR spectroscopy of metabolic disorders. Neuroimaging Clin N Am 2006;16:87-116; used with permission. Data from The Johns Hopkins University, Online Mendelian Inheritance in Man (OMIM), McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and the National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. Available at http://www.ncbi.nlm.nih.gov/omim/. Accessed October 2012. Table 33-6 Adapted From Cecil KM. MR spectroscopy of metabolic disorders. Neuroimaging Clin N Am 2006;16:87-116; used with permission. Data from The Johns Hopkins University, Online Mendelian Inheritance in Man (OMIM), McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and the National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. Available at http://www.ncbi.nlm.nih.gov/omim/. Accessed October 2012. Table 33-7 OMIM, Online Mendelian Inheritance in Man Database. Adapted From Cecil KM. MR spectroscopy of metabolic disorders. Neuroimaging Clin N Am 2006;16:87-116; used with permission. Data from The Johns Hopkins University, Online Mendelian Inheritance in Man (OMIM), McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and the National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. Available at http://www.ncbi.nlm.nih.gov/omim/. Accessed October 2012. Table 33-8 OMIM, Online Mendelian Inheritance in Man Database. Adapted From Cecil KM. MR spectroscopy of metabolic disorders. Neuroimaging Clin N Am 2006;16:87-116; used with permission. Data from The Johns Hopkins University, Online Mendelian Inheritance in Man (OMIM), McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and the National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. Available at http://www.ncbi.nlm.nih.gov/omim/. Accessed October 2012. Table 33-9 Adapted From Cecil KM. MR spectroscopy of metabolic disorders. Neuroimaging Clin N Am 2006;16:87-116; used with permission. Data from The Johns Hopkins University, Online Mendelian Inheritance in Man (OMIM), McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and the National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. Available at http://www.ncbi.nlm.nih.gov/omim/. Accessed October 2012. Table 33-10 OMIM, Online Mendelian Inheritance in Man Database. Adapted From Cecil KM. MR spectroscopy of metabolic disorders. Neuroimaging Clin N Am 2006;16:87-116; used with permission. Data from The Johns Hopkins University, Online Mendelian Inheritance in Man (OMIM), McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and the National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. Available at http://www.ncbi.nlm.nih.gov/omim/. Accessed October 2012. Three types of GM1 gangliosidosis exist: type I (infantile), type II (late infantile/juvenile), and type III (adult). An intermediate form between infantile and juvenile has been reported.1 The clinical presentation of GM1 gangliosidosis typically occurs in infancy; its features include seizures, decerebrate posturing, pitting edema of the face, hypotonia, developmental delay, hepatosplenomegaly, macrocephaly, and cherry red spots involving the macula of the retina. Additional features include broad digits, kyphoscoliosis, skeletal dysplasia with widening of the metabphyses, and dermal pigmentary lesions.2 The course is progressive, with death common within 2 years of life. A juvenile form presents during the second year of life with progressive ataxia but without many features of the infantile variety. The underlying deficiency of β-galactosidase results in accumulation of GM1 ganglioside in both gray and white matter of the cerebrum, brainstem, cerebellum, and spinal cord. Results of cerebral MRI initially are normal, with subsequent loss of cortical gray matter. Secondary changes to the white matter manifest later and typically exhibit an abnormal, nonspecific, patchy, hyperintense T2 signal within the centrum semiovale. Hypointensity of the thalami on T2-weighted images also has been reported.3 The most common forms of GM2 gangliosidoses include Tay-Sachs disease and Sandhoff disease. Tay-Sachs disease arises with β-N-acetylhexosaminidase-A isoenzyme deficiency in Jewish children of eastern European descent. Onset is usually before the age of 1 year with irritability, hypotonia, seizures, blindness, and cherry-red spots on the macula in 90% of patients. Death usually results by 2 to 3 years of age. Sandhoff disease is attributed to a deficiency of A and B isoenzymes of hexosaminidase. The clinical course is similar to that of Tay-Sachs disease. There is visceral involvement, including hepatomegaly and cardiac and renal tubular abnormalities. Brain MRI in the early stages demonstrates increased T2 signal in the basal ganglia, particularly with the enlarged caudate nuclei. Later, cortical and deep gray matter volume loss occurs with patchy increases in T2 signal in the white matter. Thalamic involvement is more reflective of Sandhoff disease. In adult-onset Sandhoff disease, lower motor neuron involvement has been reported. Whereas cerebellar atrophy may vary, it does not appear to be correlated with clinical severity.4 In persons with Tay-Sachs disease, the thalami may be hypointense on T2-weighted images and hyperintense on T1-weighted images because of calcium deposition. It has been reported that T2-weighted hyperintensity in cerebral matter is indicative of abnormal myelin production and active demyelination.5 Asymmetrical swelling and high T2 intensity in the white matter and basal nuclei of the right hemisphere has been reported. Elevated levels of cytokines have been reported, possibly indicating inflammation as a contributing factor to the progression of gangliosidosis.6 In the B1 variant of GM2 gangliosidosis, the bilateral thalami may appear hyperdense/hyperintense on CT/T1-weighted MRI and show a T2-hypointense signal in the ventral thalami and a hyperintense signal in the posteromedial thalami. Other findings in this variant include involvement of the medullary lamellae, bilateral T2 hyperintense/swollen basal ganglia, diffuse white matter hyperintensity on T2-weighted images, and brain atrophy in later stages.7 Cerebral MRI demonstrates two major abnormalities: 1. Diffuse patches of hyperintensity on T2-weighted images resulting from accumulation of mucopolysaccharide in neurons and astrocytes and degenerative effects on myelin 2. Prominent cystic or perivascular spaces resulting from metabolic accumulation within histiocytes located in perivascular areas, which is the most characteristic imaging feature; reversal of these changes after bone marrow transplantation has been reported Spinal stenosis is especially common in Morquio and Maroteaux-Lamy variants but may be demonstrated in other variants. Typical bullet-shaped vertebral bodies are characteristic (Fig. 33-1). Figure 33-1 A 2-year-old boy with Hunter syndrome, mucopolysaccharidoses, type II. NCL is a disorder or group of disorders characterized by striking volume loss of brain parenchyma. NCL, which can be divided into six subtypes based on age at onset, is one of the most common neurodegenerative syndromes, with an incidence of 1 per 25,000 live births. The various subtypes are associated with different mutations in the CLN genes and have similar clinical manifestations occurring at different ages. These manifestations include seizures and abnormal eye movements, with subsequent vision loss, dementia, hypotonia, and speech and motor deficits. CSF neurotransmitter abnormalities also have been reported in patients with NCL.8 At pathology, these disorders are characterized by distinctive granular inclusions in neuronal lysosomes, called granular osmiophilic deposits. Imaging findings follow behind the clinical presentation in all but the infantile form of NCL and are dominated by progressive cerebral and cerebellar volume loss. Later stages of disease are characterized by development of a band of hyperintense signal in the periventricular white matter on T2-weighted images. In palmitoyl protein thioesterase-1 related NCL, isolated, symmetric dentate nucleus hyperintensities have been reported in early stages on T2-weighted images.9 Proton MRS has shown progressive decreases in NAA and relative increases in mI in persons with NCL. In persons with MLD, the primary metabolic defect is a deficiency in the enzyme arylsulfatase A, resulting in the accumulation of cerebroside sulfate within the lysosome. MLD has four subtypes: congenital, late infantile, juvenile, and adult. The late infantile subtype is the most common and presents from around 14 months to 4 years of age. The early presentations are an unsteady gait that progresses to severe ataxia and flaccid paralysis, dysarthria, mental retardation, and decerebrate posturing. Gallbladder involvement has been reported, possibly appearing before the onset of neurologic symptoms. Intestinal involvement also has been reported, specifically polypoid masses in one patient.10 Histologic analysis of the abnormal nervous tissue demonstrates a complete loss of myelin (demyelination) followed by axonal degeneration. Metachromatic granules are reported within engorged lysosomes in white matter and neurons and on peripheral nerve biopsies. Oligodendrocytes are reduced in number, and areas of demyelination predominate throughout the deep white matter region. Early sparing of the subcortical arcuate white matter fibers (“U” fibers) occurs until late in the disease process. An inflammatory response typically is absent, which accounts for a lack of enhancement in this disorder, but eventually, even myelinated white matter is replaced by astrogliosis and scarring. The corpus callosum is involved before significant progression, whereas subcortical white matter remains unaffected until the disease has progressed; atrophy is a late sign. Demyelination also can be seen in the posterior limbs of the internal capsule, descending pyramidal tracts, and the cerebellar white matter.11 Thalamic changes may be common in primary MLD, and isolated cerebellar atrophy may be seen in some atypical later-onset variants. On T2-weighted images, there is marked hyperintensity of the white matter fiber tracts involving the cerebral hemispheres that may extend to the cerebellum, brainstem, and spinal cord. The findings further demonstrate diffuse deep white matter involvement with relative sparing of subcortical white matter. The findings initially are focal and patchy, but later, a diffuse, hyperintense T2 signal of the centrum semiovale develops. Two distinct white matter appearances have been noted that mimic what was previously considered to be pathognomonic of Pelizaeus-Merzbacher disease (PMD). Punctate areas of hypointensity (“leopard skin” appearance) and radiating patterns of linear tubular structures of T2 hypointensity (“tigroid” appearance) are seen, with areas of relatively normal-appearing white matter within the areas of demyelination. On T1-weighted images, the white matter fibers may be isointense with, or hypotense to, gray matter (Fig. 33-2). Figure 33-2 A 7-year-old boy with metachromatic leukodystrophy. Proton MRS studies have demonstrated reduced NAA, which is expected with neuroaxonal loss, but they also have revealed disturbances in glial cell metabolism associated with elevated mI and choline. The levels of NAA in white matter have been found to correlate with motor function in children with MLD.12 Globoid cell leukodystrophy (Krabbe disease) arises from a deficiency in the enzyme β-galactocerebrosidase, leading to the accumulation of cerebroside and galactosylsphingosine, which induces apoptosis in the oligodendrocyte cell lines. Globoid cell leukodystrophy, an autosomal-recessive disorder, has a frequency of 2 in 100,000 in a series reported from Sweden. It is seen predominantly in young children; however, the infantile form is the most common. Onset of symptoms usually begins between 3 and 5 months after birth with irritability. The disease continues to progress, with development of symptoms mimicking encephalitis with motor deterioration and atypical seizures. At the end stage of the disease, the child is in a vegetative state with decerebrate posturing. Elevated CSF protein has been reported, to a larger extent in adult phenotypes than in phenotypes affecting younger people.13 Positional ocular flutter has been reported in one patient with infantile Krabbe disease.14 In nerve conduction studies, the severity of abnormalities appears to correlate with the severity of clinical symptoms.15 Delayed myelination may be the first finding noted on MRI in infants with this disorder. In infantile Krabbe disease, MRI findings may be normal, but as the disease progresses, classic Krabbe features emerge; this phenomenon is probably related to the immature myelination.16 The appearance of Krabbe disease on MRI is featured as one of either two patterns. A patchy hyperintense periventricular signal on T2-weighted images, consistent with hypomyelination, eventually may evolve into a more diffuse pattern in the white matter. In this form, involvement of the thalami with a hyperintense T2 signal often is present as well. A second pattern is a patchy low signal on T2-weighted images in a similar distribution to the hyperdense regions seen on CT, which is suspected to represent a paramagnetic effect from calcium deposition in the region. Additional early changes include increased density in the distribution of the thalami, cerebellum, caudate heads, and brainstem that may precede the abnormally low attenuation of white matter in the centrum semiovale. Symmetric enlargement of the optic nerves also has been described in persons with Krabbe disease, which is presumed to reflect accumulation of proteolipid in globoid cells. The distal optic nerves are primarily involved; however, a case has been described with proximal prechiasmatic enlargement of the nerves.17 At times, changes within the cerebellar white matter also have been reported, with hyperintensity on T2-weighted images. The findings within the spinal cord are visualized as atrophic changes. Diffuse volume loss and periventricular white matter abnormalities predominate in the latter stages of this disease (Fig. 33-3). Figure 33-3 A 4-year-old girl with Krabbe disease.
Inherited Metabolic and Neurodegenerative Disorders
Inherited Metabolic Brain Disorders
Lysosomal Storage Diseases
Gangliosidoses
GM1 Gangliosidosis
GM2 Gangliosidosis
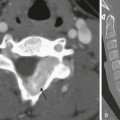
Mucopolysaccharidoses
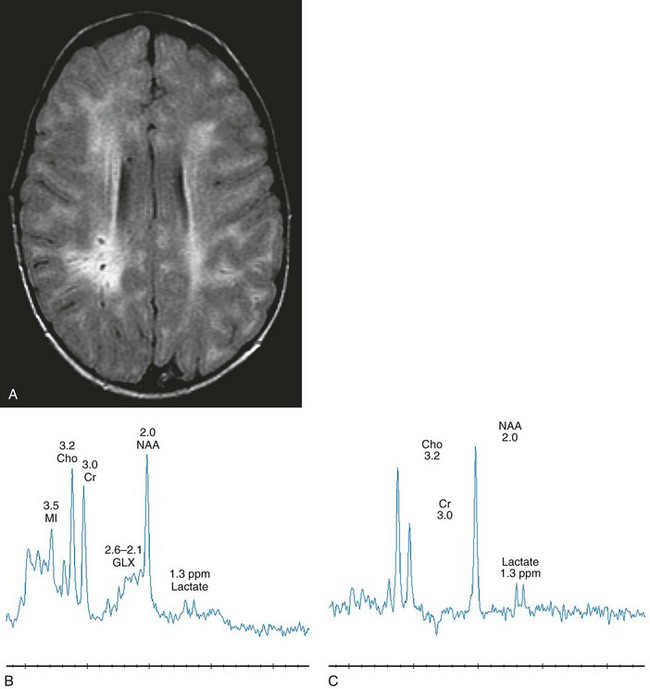
Select axial fluid-attenuated inversion recovery (A), short echo (time to echo [TE] 35 msec) magnetic resonance spectroscopy (MRS) (B), and long echo (TE 288 msec) MRS (C) images are shown. The patient underwent scanning within months of a stem cell transplant. Prominent perivascular spaces are demonstrated with a diffuse, abnormal, hyperintense signal throughout the periventricular and subcortical white matter. The spectra demonstrate diminished N-acetylaspartate levels with elevated lactate, which may reflect histiocytic cell infiltration of the perivascular spaces and brain parenchyma. (From Cecil KM. MR spectroscopy of metabolic disorders. Neuroimaging Clin N Am. 2006;16:87-116; used with permission.)
Neuronal Ceroid Lipofuscinoses
Metachromatic Leukodystrophy
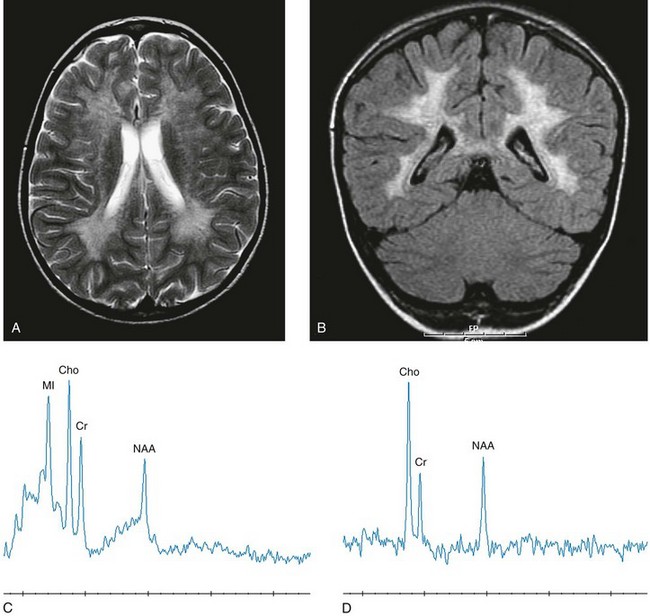
Select axial T2-weighted (A), coronal fluid-attenuated inversion recovery (B) with left parietal white matter short echo magnetic resonance spectroscopy (MRS) (C) and long echo MRS (D) images reveal an abnormal hyperintense signal in the periventricular white matter throughout the cerebrum, sparing the subcortical U fibers. The signal has a “tigroid” appearance on T2-weighted images. The spectra demonstrate significant elevations of choline and myo-inositol with diminished N-acetylaspartate reflecting neuroaxonal loss, demyelination, and glial activation. (From Cecil KM. MR spectroscopy of metabolic disorders. Neuroimaging Clin N Am. 2006;16:87-116; used with permission.)
Globoid Cell Leukodystrophy (Krabbe Disease)
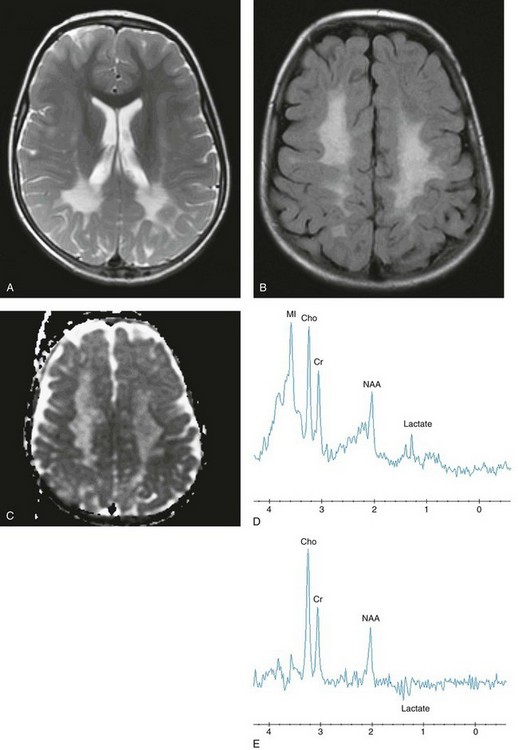
Select axial T2-weighted fast spin echo (A), axial fluid-attenuated inversion recovery (FLAIR) (B), axial apparent diffusion coefficient (ADC) map (C), short echo magnetic resonance spectroscopy (MRS) (D), and intermediate (time to echo 144 msec) echo MRS (E) images are shown. Abnormal hyperintense T2 and FLAIR signals are noted within the centrum semiovale with sparing of the subcortical U fibers and posterior limbs of the internal capsules. Relatively increased diffusion is seen in the periatrial and parietal white matter. The spectra demonstrate significant elevations of lactate, choline, and myo-inositol, with diminished N-acetylaspartate reflecting neuroaxonal loss, demyelination, and glial activation.
Radiology Key
Fastest Radiology Insight Engine