CHAPTER 32 Intra-Axial Neoplasms
Primary neoplasms of the central nervous system (CNS) are relatively infrequent, accounting for 16.5 to 18 cases per 100,000 person-years. However, they are a significant health concern.
Primary CNS neoplasms are histologically diverse and may arise from neuroepithelial tissue (e.g., astrocytic tumors, oligodendroglial tumors, ependymal tumors, embryonal tumors) or the hematopoietic system (primary CNS lymphoma). Neuroepithelial tissue consists of glial cells, neuronal cells, neuroblastic cells, pineal parenchymal cells, and residual embryonal cells. Most intra-axial neoplasms are of glial origin, accounting for 40% to 50% of primary CNS neoplasms.1 The glial neoplasms arise from astrocytes, oligodendrocytes, ependymal cells, or their derivatives in the choroid plexus. The ganglion cell tumors (ganglioglioma, gangliocytoma) are composed of abnormal neoplastic neuronal cells or of a combination of neuronal and astrocytic elements.
Primary CNS neoplasms are graded according to the World Health Organization (WHO) classification system. The high-grade glial neoplasms are differentiated from the low grade on pathology by the presence of increased cellularity, increased mitotic activity, necrosis, and vascular proliferation. In a surgical series of stereotactic brain biopsies—consisting of 5000 specimens—the most common intra-axial brain masses were high-grade primary neoplasms (36%), low-grade primary neoplasms (33%), metastasis (8%), lymphoma (5%), demyelinating inflammatory lesions (3%), infarcts (2%), and abscesses (1%).2,3
Imaging plays an integral role in the detection, diagnosis, and management of these lesions, and advanced imaging, such as perfusion and MR spectroscopy, may help narrow the differential diagnosis and assist in post-treatment follow-up. However, the limitations of conventional MRI and advanced imaging must be kept in mind when interpreting the results. Conventional MRI is currently limited in its ability to demarcate the exact margins of infiltrative tumors, to accurately grade the neoplasm, and to monitor early changes after treatment. An example of the limitation of advanced imaging is in the evaluation of MR spectroscopy where there are no unequivocal cutoff metabolite peaks that clearly differentiate a neoplastic lesion from a non-neoplastic one, and low-grade neoplasms may have spectra that are similar to high-grade neoplasms, lymphoma, and metastases (i.e., elevation in choline [Cho], reduction in N-acetyl aspartate [NAA], and presence of a lipid/lactate peak). Tumefactive demyelinating lesions may show this pattern as well. However, in a review by Al-Okaili and associates, it was found that a Cho/NAA cutoff ratio of 2.2 could reliably separate high-grade neoplasms from low-grade neoplasms and non-neoplastic lesions.3
Perfusion-weighted imaging may be of benefit in assessing angiogenesis induced by neoplasms. The vessels formed in a neoplastic process tend to be abnormal and leaky, thus resulting in increased permeability parameters on perfusion MRI. Several studies have demonstrated a correlation of tumor blood volume with the grade of neoplasm.4–6 Law and colleagues observed that a relative cerebral blood volume (rCBV) threshold value of 1.75 provided a sensitivity of 95% and positive predictive values of 87% for distinguishing between high- and low-grade gliomas.4 Because metastases tend to induce angiogenesis as well, the perfusion parameters of metastatic lesions tend to overlap with high-grade neoplasms.
Recent research has demonstrated that diffusion tensor imaging (DTI) has important potential benefits in surgical planning owing to the ability to detect white matter tracts in the region of the neoplasm.7,8 The ability to assess whether the neoplasm has shifted the tracts versus infiltrating them is important information for the neurosurgeon and can assist in determining the method of approach and the extent of resection that is possible.
PILOCYTIC ASTROCYTOMA
Epidemiology
Pilocytic astrocytomas are the most common form of glioma in childhood and most frequently develop in the first two decades of life; 80% occur in patients younger than 20 years old.9 Rarely, they arise in patients older than 50. They comprise 5% to 10% of all gliomas, and there is no strong gender predilection. However, some series have reported a slightly higher incidence in females (11 : 9). Pilocytic astrocytoma may arise anywhere within the neuraxis, but within the pediatric population (<15 yr) they arise more frequently infratentorially and comprise 85% of all cerebellar neoplasms in the pediatric age group.9 Other common sites are the optic nerve, optic chiasm and hypothalamus, basal ganglia, and thalamus.
Pilocytic astrocytomas are associated with neurofibromatosis type 1 (NF-1), occurring in 15% to 21% of patients with this disease.9 In these patients the intraconal optic nerve and chiasm are most commonly involved (Fig. 32-1). The majority of optic pathway gliomas occur before 6 years of age, and there is a female predominance (2 : 1).
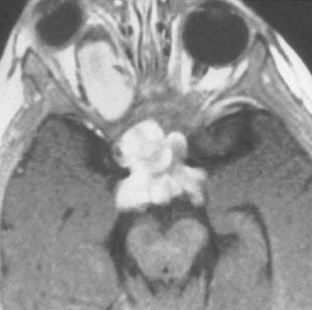
FIGURE 32-1 Optic nerve glioma. Axial T1W postcontrast MR image from a patient with neurofibromatosis type 1 demonstrates an enhancing mass involving the optic nerve and chiasm.
Pilocytic astrocytomas have benign biologic behavior with a survival rate of up to 94% at 10 years.9 Disseminated disease and recurrence are very rare. Interestingly, when metastatic disease occurs, it can do so without increased mortality, unlike metastases from higher-grade neoplasms (Fig. 32-2).
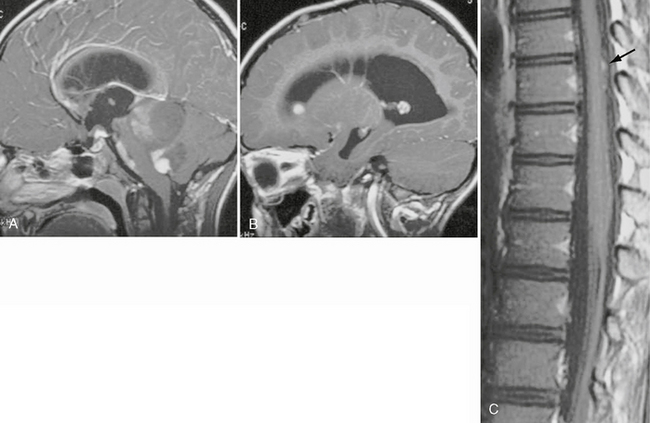
FIGURE 32-2 Disseminated pilocytic astrocytoma. A, Sagittal postcontrast T1W MR image demonstrates a cystic lesion with an enhancing nodule in the posterior fossa. B, Sagittal postcontrast T1W MR image demonstrates an enhancing nodule in the anterior aspect of the lateral ventricle. C, Sagittal postcontrast T1W MR image of the spine reveals enhancing nodules (arrow) along the posterior aspect of the spinal cord.
Pathophysiology
Pilocytic astrocytomas are associated with NF-1, and approximately 15% of patients with NF-1 will develop a pilocytic astrocytoma, most commonly of the optic nerve. Pilocytic astrocytomas are a major source of morbidity in patients with NF-1. Cytogenetic studies of sporadic pilocytic astrocytomas have demonstrated a loss of genetic material of the long arm of chromosome 17(17q), which is near the same locus as the NF1 tumor suppressor gene.10 Other chromosomal mutations have been found on chromosomes 7, 8, 11, 19, and 21.11
Pilocytic astrocytomas typically maintain their WHO grade I histology, tending toward degenerative changes over time rather than evolving into a more aggressive lesion. Rarely, progression to a malignant lesion has been reported; however, these tumors tend to have a better prognosis than glioblastoma multiforme and are referred to as anaplastic pilocytic astrocytoma. Most of the pilocytic astrocytomas that underwent malignant change had previously been irradiated, and it is possible that radiation may have been a factor in the malignant degeneration. Occasionally, pilocytic astrocytomas may seed the cerebrospinal fluid (CSF) and neuraxis, and in these cases the hypothalamus is usually the primary site rather than the cerebellum.
Pathology
On gross pathology, pilocytic astrocytomas are soft, wellcircumscribed lesions. Fluid accumulation (“cyst formation”) is commonly seen (Fig. 32-3). In older lesions, hemosiderin or calcium may be seen. Calcification tends to occur more often in tumors arising from the optic nerve or hypothalamic-thalamic regions. Lesions involving the optic nerve result in elongation and fusiform widening of the nerve.
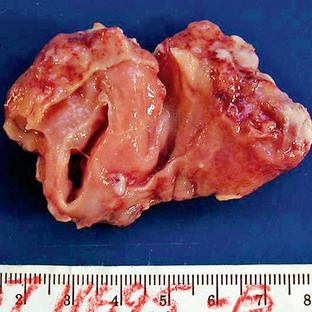
FIGURE 32-3 Pilocytic astrocytoma. Bisected gross specimen reveals a soft, gelatinous, pink cystic mass.
Pilocytic astrocytomas have increased capillary vascularity and low to moderate cellularity. In long-standing neoplasms the vessels may become hyalinized and glomeruloid. A biphasic pattern of two astrocyte populations having varying proportions of compact bipolar cells with Rosenthal fibers (nonfilamentous electron-dense masses) and loose-textured multipolar cells with microcysts and eosinophilic granular bodies/hyaline droplets is seen (Figs. 32-4 and 32-5). The presence of eosinophilic granular bodies is thought to indicate slow growth and low histologic grade and is associated with an improved prognosis. Neoplastic changes involving the cyst wall have been reported.12 Infiltration into the surrounding tissues may be seen but is usually shallow, especially in comparison with the adult or diffuse astrocytomas. This occurs more frequently in tumors involving the optic nerve and chiasm, and, commonly, there is poor demarcation between tumor and normal tissue. Pilocytic astrocytomas involving the cerebellum may infiltrate the leptomeninges, resulting in fixation of the cerebellar folia and filling of the sulci (Fig. 32-6).
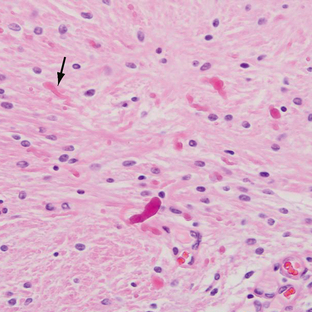
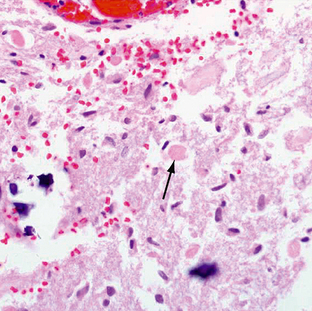
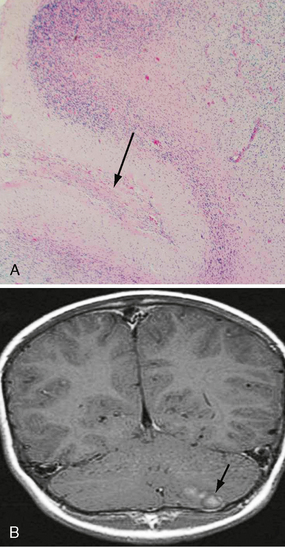
FIGURE 32-6 Pilocytic astrocytoma. A, H&E stain demonstrates the pilocytic astrocytoma “filling in” the subarachnoid space (arrow). B, Corresponding coronal postcontrast T1W MR image demonstrates an enhancing lesion in the inferior aspect of the left cerebellar hemisphere.
Imaging
Pilocytic astrocytomas are well-circumscribed lesions that enhance on postcontrast imaging. Fluid formation is common, and the classic description is that of a “cyst and nodule” (Fig. 32-7). Occasionally, calcification (up to 25%) may be seen and hemorrhage has been reported (Figs. 32-8 and 32-9). Pilocytic astrocytomas occasionally may enhance in a ring-like pattern, suggesting the morphology of a higher-grade neoplasm. In these cases, location (cerebellum) and age (<15 years) may suggest benign fluid, rather than necrosis, as the cause of the ring enhancement.
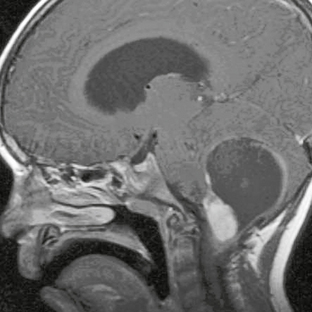
FIGURE 32-7 Pilocytic astrocytoma. Sagittal postcontrast T1W MR image demonstrates the classic “cyst and nodule” appearance of a pilocytic astrocytoma in the posterior fossa. Thin rim enhancement is seen involving the cyst wall.
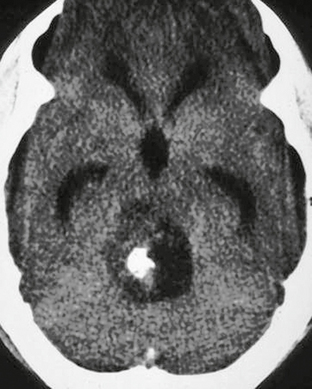
FIGURE 32-8 Pilocytic astrocytoma. Noncontrast CT demonstrates a hypoattenuating lesion in the posterior fossa. A focus of high attenuation is present consistent with calcification.
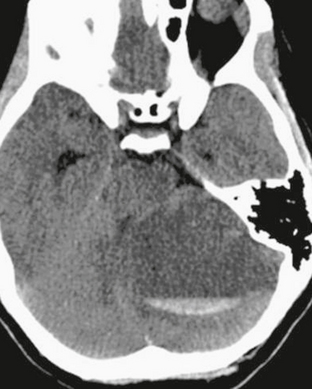
FIGURE 32-9 Pilocytic astrocytoma. A low-attenuation mass lesion is present within the left cerebellar hemisphere consistent with a cyst, which has a fluid-fluid level consistent with hemorrhage.
Four imaging patterns of pilocytic astrocytomas have been described: (1) enhancing mural nodule and nonenhancing cyst, (2) enhancing cyst wall and enhancing mural nodule, (3) “necrotic” mass with central nonenhancing region, and (4) predominantly solid mass with minimal or no cyst.12 Cyst wall enhancement does not necessarily imply there is tumor involvement, and removal of the cyst wall is not related to improved survival.13,14 Some pilocytic astrocytomas may show thin rim enhancement from reactive gliosis surrounding the fluid.
CSF dissemination is rare (2%-12%) and increases in the setting of tumors located in the hypothalamus, in cases of partial resection, and in patients younger than 4 years old at the time of diagnosis. Dissemination tends to occur within the first 3 years after diagnosis.15
MRI
On MRI, the solid portion of the neoplasm is typically isointense to hypointense on T1-weighted (T1W) imaging and hyperintense on T2-weighted (T2W) imaging to gray matter. The cystic portion is often hyperintense to CSF from protein (Fig. 32-10). The cyst contents do not suppress on FLAIR. The solid portion enhances on postcontrast imaging. Occasionally, the wall around the fluid may demonstrate rim enhancement. Rarely, leptomeningeal metastases may be seen.
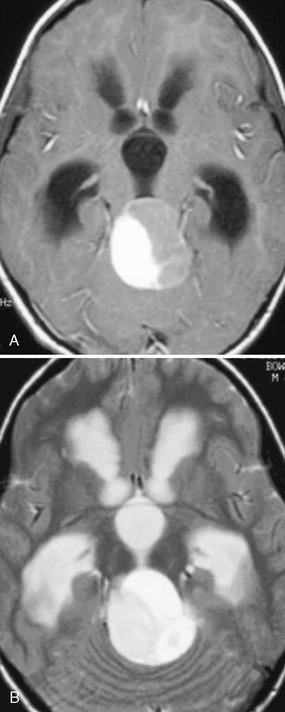
FIGURE 32-10 Pilocytic astrocytoma. A, Axial postcontrast T1W MR image demonstrates a cyst with an enhancing mural nodule. B, T2W imaging demonstrates that the fluid within the cystic portion of the lesion is hyperintense to CSF. Hydrocephalus is present.
In these low-grade tumors, MR spectroscopy demonstrates elevation in choline and a reduction in NAA, a pattern that is also seen in higher-grade neoplasms. There is minimal elevation in the lipid peak, and lactate peaks are elevated (Fig. 32-11). The elevation in lactate may represent alterations in mitochondrial metabolism or represent variability in glucose utilization rates among low-grade astrocytomas and not necrosis because necrosis is rare in pilocytic astrocytomas.16
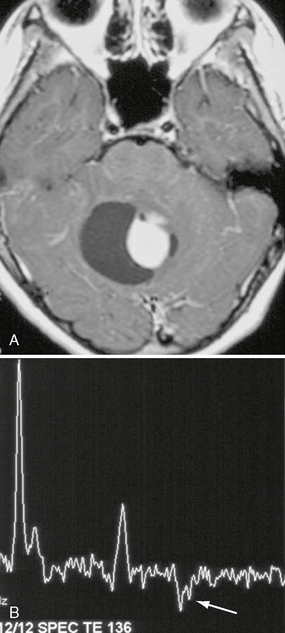
FIGURE 32-11 Pilocytic astrocytoma. A, Axial postcontrast T1W MR image demonstrates a cyst with an enhancing mural nodule. B, Single-voxel MR spectroscopy (TE = 136) placed in the region of the enhancing nodule demonstrates a more ominous-appearing MR spectroscopy pattern than would be expected for a grade I lesion. Choline level is elevated, NAA level is reduced, and lactate is present (arrow).
PILOMYXOID ASTROCYTOMA
Pilomyxoid astrocytoma is a WHO grade II neoplasm that is closely related to pilocytic astrocytoma.
Clinical Presentation
Pilomyxoid astrocytomas have nonspecific symptoms related to their location. They can occur anywhere along the neuraxis but have a predilection for the hypothalamus. The most common presenting symptoms are those related to increased intracranial pressure or parenchymal compression. Other reported symptoms include developmental delay, failure to thrive, altered level of consciousness, feeding difficulties, and generalized weakness. Pilomyxoid astrocytomas behave more aggressively than pilocytic astrocytomas, and they have a shorter progression-free and overall survival. They also have a higher rate of recurrence and CNS dissemination.17
Pathophysiology
These neoplasms behave more aggressively than pilocytic astrocytomas, with local recurrence (76% for pilomyxoid astrocytomas compared with 50% for pilocytic tumors) occurring more frequently, despite having equal rates for gross total resection. CSF dissemination is relatively common, occurring in up to 14%.18 In addition, 33% of patients with pilomyxoid astrocytoma died of their disease, compared with only 17% of patients with pilocytic astrocytoma.18
Pathology
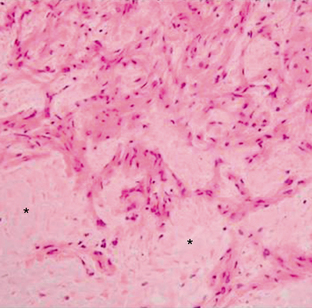
Pilomyxoid astrocytomas are described as soft gelatinous masses. They have a prominent mucoid matrix and an angiocentric arrangement of bipolar tumor cells, resembling the perivascular rosettes seen in ependymomas (Fig. 32-12). Monomorphous piloid cells are present in a loose fibrillary and myxoid background. They do not demonstrate the Rosenthal fibers or eosinophilic granular bodies that are common in pilocytic astrocytomas. Rare mitotic figures may be seen. In some cases, infiltration of tumor cells into the surrounding neuropil occurs at the periphery of the neoplasm.19
Imaging
Pilomyxoid astrocytomas may occur anywhere along the neuraxis but are most common in the hypothalamic/chiasmatic region (76.9%).20 On imaging, pilomyxoid astrocytomas are predominantly solid neoplasms, in contrast to the “cyst and nodule” frequently seen in pilocytic astrocytomas (Fig. 32-13). Pilomyxoid astrocytomas may occasionally demonstrate a minimal cystic component. On postcontrast imaging, pilomyxoid astrocytomas have been described as demonstrating homogeneous enhancement, although the number of cases in the literature is limited. Hydrocephalus is frequently present. Hemorrhage is rare, with only a few cases reported.21,22
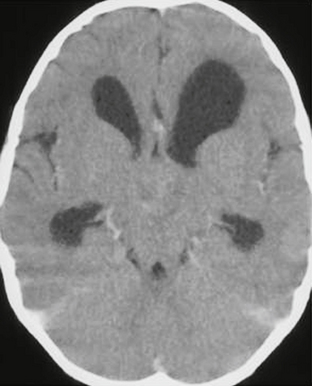
FIGURE 32-13 Pilomyxoid astrocytoma. Contrast-enhanced CT demonstrates a minimally enhancing, solid, hypothalamic lesion. Hydrocephalus is present.
PLEOMORPHIC XANTHOASTROCYTOMA
Epidemiology
PXAs typically occur in children and young adults, with two thirds occurring before the age of 18 years; however, there have been rare reports of the lesions in older adults (age range: 5-82 years). These are uncommon neoplasms, accounting for less than 1% of astrocytic neoplasms.1 There is no documented gender predilection. The majority occur in the supratentorial brain (98%), most commonly the temporal lobe (49%).1
Pathophysiology
PXAs are occasionally associated with cortical dysplasia or with ganglionic lesions, suggesting that their formation may be promoted in malformative states. They have also been reported in patients with NF1, although no definite association with hereditary tumor syndromes has been demonstrated. Because of their typical superficial location, origin from subpial astrocytes is postulated. However, the histogenesis is unclear and there is the suggestion that this tumor may derive from bipotential precursor cells.23
Long-term survival in patients with PXAs is reported at 81% at 5 years and 70% at 10 years.24 The degree of resection appears to be the best predictor of outcome, along with a low mitotic index. The tumor has a relatively high rate of recurrence; a malignant transformation occurs in up to 20%.24
Pathology
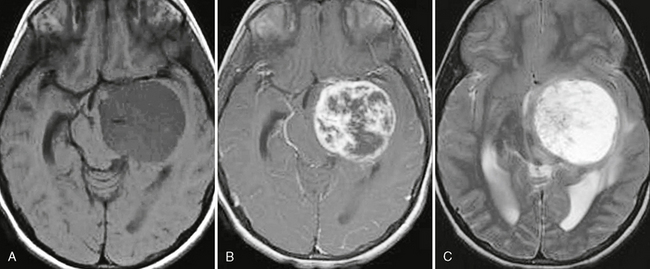
PXAs are peripherally located neoplasms and involve the leptomeninges (Fig. 32-15). Cyst formation is common, and a “cyst and nodule” appearance is common.
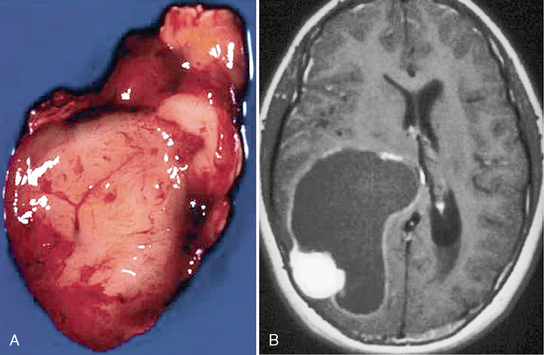
FIGURE 32-15 Pleomorphic xanthoastrocytoma. A, Gross specimen of the solid nodular component demonstrates a seemingly well-demarcated lesion. B, Corresponding postcontrast T1W MR image reveals a cystic lesion with the enhancing mural nodule. The cystic lining enhances as well.
Pleomorphic xanthoastrocytomas have a variable histologic appearance, hence the name “pleomorphic.” Mononucleated or multinucleated giant astrocytes with variable nuclear size and staining are seen. They have a dense reticulin network and lipid (xanthomatous) deposits within the tumor cells. The term xanthoastrocytoma refers to large cells with intracellular lipid accumulation (Fig. 32-16). Despite a circumscribed appearance, most PXAs demonstrate extension into the surrounding brain. Even though the tumor is frequently attached to the leptomeninges, dural invasion is uncommon. Necrosis within the tumor portends a poorer prognosis.25 Mitoses are usually absent or rare; however, atypical PXAs demonstrate high mitotic activity and marked hypercellularity and necrosis. These may be considered grade III or anaplastic PXA.
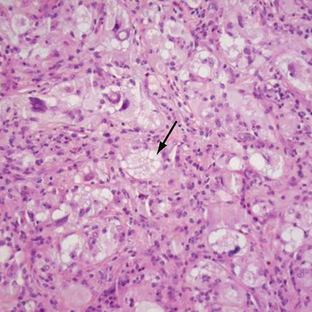
FIGURE 32-16 Pleomorphic xanthoastrocytoma. Xanthic cells, expressed as vacuolization of the neoplastic cells, are shown (arrow).
Immunohistochemical analysis demonstrates the presence of glial fibrillary acidic protein (GFAP) and S-100 (present in cells derived from the neural crest); and neuronal markers such as synaptophysin (protein present in neurons in the brain and spinal cord that participate in synaptic transmission), class III β-tubulin (a neuronal marker), and NF proteins (play an important role in neuronal development) are also described. TP53 is the tumor suppressor gene, and mutations occur in many human cancers. Analysis for the TP53 mutation is negative or only focally positive. In one study of PXAs that demonstrated malignant progression, histochemical analysis revealed GFAP positivity in 100% and S-100 and TP53 were expressed in 67% but synaptophysin and NF protein were absent.26 The MIB-1 index for PXAs is typically less than 1%.
Imaging
The classic imaging appearance of PXAs is that of a peripherally located “cystic” supratentorial mass. However, 52% do not have macroscopic fluid changes.1 Lesions may be well circumscribed or poorly defined.
CT
On CT, the solid portion of the PXA is hyperattenuating and the cystic portion is hypoattenuating. The solid portion enhances on postcontrast imaging. Calcification is rare (Fig. 32-17). Overlying skull remodeling, erosion, or lytic change is uncommon; when present it suggests slow progression of the tumor.
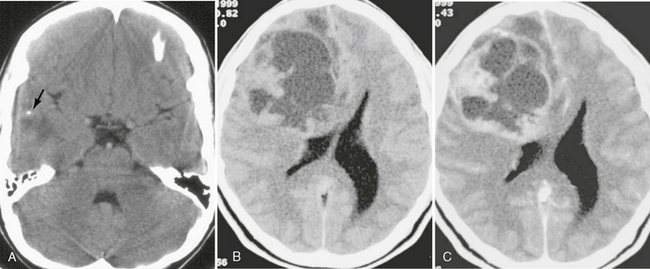
FIGURE 32-17 Pleomorphic xanthoastrocytoma. A, Noncontrast CT demonstrates a mixed attenuation lesion in the right temporal lobe. A small focus of calcification is seen (arrow). B, Noncontrast CT from another patient demonstrates a large cystic and solid mass involving the right frontal lobe. C, Postcontrast CT demonstrates an enhancing nodule and enhancement of the cyst walls.
MRI
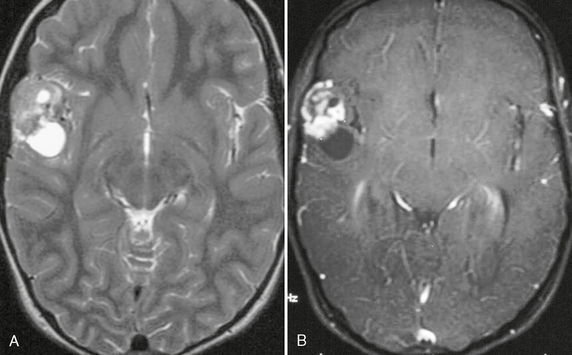
The solid portion of a PXA is isointense to gray matter on T1W and T2W images. Peritumoral edema may be seen, but it is uncommon. On postcontrast imaging the solid portion enhances (Fig. 32-18). Involvement of the leptomeninges is highly characteristic of PXA, and enhancement along the adjacent meningeal surface may be seen.
DIFFUSE ASTROCYTOMA
Clinical Presentation
These lesions may grow slowly over several years, but over time almost all will progress to higher-grade gliomas (WHO grade III or IV). The management of these patients is debated. Some centers treat them aggressively; however, studies have not produced evidence of improved survival after radical surgery or early radiation therapy. Other centers have adopted a “watch and wait” policy with clinical and imaging surveillance.27–29
Pathophysiology
A genetic hallmark for these neoplasms is frequent TP53 mutation, which is seen in greater than 60%. TP53 is the gene that codes for the TP53 protein, an essential transcription factor. TP53 mutations in grade II astrocytoma may be seen in patients with Li-Fraumeni syndrome, although these patients typically develop higher-grade neoplasms (III or IV). An association with inherited multiple enchondromatosis type 1 (Ollier disease) has also been noted. Overexpression of platelet-derived growth receptor-α is also reported. Chromosomal abnormalities are also reported (e.g., gain of 7q, 8q amplification, LOH 10p, 22q, chromosome 6 deletions).
Pathology
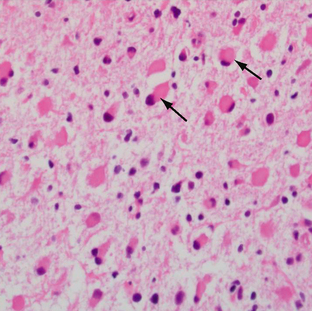
The fibrillary astrocytoma is the most common histologic subtype of diffuse astrocytoma. Gemistocytic (from the Greek gemisto meaning “stuffed”) is the other common subtype, and it has a higher and more rapid rate of progression to glioblastoma (Fig. 32-19). Overall, the infiltrated white matter shows increased cellularity in comparison with the normal brain and occasional nuclear atypia is present. Mitotic activity is typically absent in grade II astrocytoma. No necrosis or neovascular proliferation is present. Infiltration of adjacent structures is seen. On immunohistochemistry, GFAP and S-100 protein are positive. The MIB-1 index is low, correlating with the low mitotic activity.
Imaging
CT
Diffuse astrocytomas are typically isoattenuating to hypoattenuating masses. They do not enhance on postcontrast imaging and, when enhancement is present on follow-up examinations, suspicion should be raised for transformation to a higher-grade neoplasm. Calcification occurs in approximately 20%, and macroscopic cyst formation is rare.
MRI
Diffuse astrocytomas are low in signal on T1W imaging and may expand the white matter and adjacent cortex. They may cause loss of signal contrast between gray and white matter. On T2W imaging they are hypointense to hyperintense and may appear circumscribed. This is deceptive, because pathologically they are not discrete. Surrounding or spreading vasogenic edema is uncommon, and calcification, cysts, and hemorrhage are rare. They do not enhance on postcontrast imaging (Fig. 32-20).
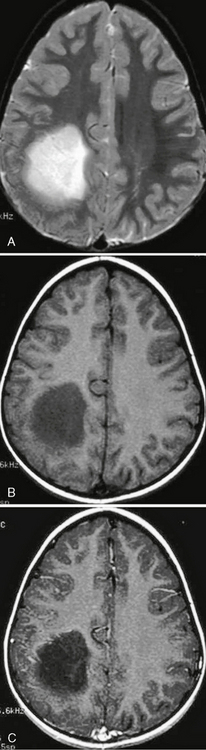
FIGURE 32-20 Grade II astrocytoma. A, Axial T2W MR image demonstrates a relatively well-circumscribed hyperintense mass in the left parietal lobe. B, The lesion is hypointense on T1W imaging and does not demonstrate enhancement on the postcontrast image (C).
MR spectroscopy should show an elevation of choline and a reduction in NAA. A high myoinositol (mI)/creatine (Cr) ratio is also present (0.82 ± 0.25).30 On dynamic contrast-enhanced T2*W the rCBV is typically low and should be lower than values seen in the high-grade astrocytomas; rCBV is typically less than 1.75.4 A study by Danchaivijitr and coworkers found that evaluation of the rCBV was able to distinguish between patients whose tumor did not transform to a higher grade and those whose tumor did over a 23-month observation period. In the nontransforming stable neoplasms the rCBV measurements remained low, whereas in those tumors that transformed, the mean rCBV value at the point of transformation was elevated to 5.36 ± 3.01. The elevated rCBV occurred before the development of contrast enhancement and may be an “early warning signal.”29
An assessment of the utility of DTI in differentiating diffuse astrocytomas from anaplastic was performed by Goebell and associates, who found that the fiber tracts were better preserved along the periphery of low-grade (II) neoplasms, whereas the tracts tended to be disarranged (lower anisotropy) in high-grade gliomas. Within the center of both low- and high-grade gliomas the fractional anisotropy (FA) was low, which was consistent with a high degree of disorganization of myelinated fiber tracts.31
ANAPLASTIC ASTROCYTOMA
Anaplastic astrocytomas are WHO grade III, diffusely infiltrating neoplasms. They are also referred to as grade III astrocytomas. Confusing synonyms include “malignant astrocytoma,” “malignant glioma,” and high-grade astrocytoma. However, these terms have also been used for glioblastoma, which is a WHO grade IV lesion.
Pathology
These tumors demonstrate increased cellularity, nuclear atypia, and significant proliferative activity (Fig. 32-21). The GFAP is positive, and the MIB-1 index is 5% to 10%. Microvascular proliferation and necrosis are absent (according to the current WHO revision of 2007).
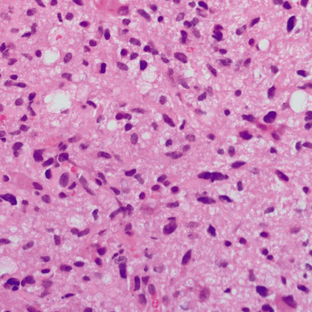
Imaging
MRI
Anaplastic astrocytomas are hypointense or isointense mass lesions located within the white matter, but they may involve the overlying cortex, resulting in its expansion. On postcontrast imaging there is usually no enhancement. T2W images reveal a heterogeneously hyperintense mass (Fig. 32-22). MR spectroscopy demonstrates a decrease in NAA and an elevation in the Cho/Cr ratio. Lower mI/Cr ratios are seen than in low-grade gliomas (0.33 ± 0.16).30 Some authors suggest that mI reflects well-differentiated astrocytic cells.
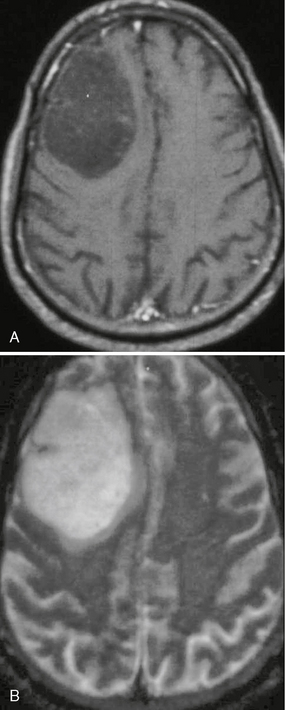
FIGURE 32-22 Anaplastic astrocytoma. A, T1W postcontrast MR image demonstrates a nonenhancing, hypointense mass lesion within the right frontal lobe. B, On T2W imaging the lesion appears hyperintense and relatively circumscribed.
Sugahara and associates found mean maximum rCBV values of 5.84 in anaplastic astrocytomas.32 Assessment with DTI reveals greater derangement of fiber tracts at the periphery of anaplastic astrocytomas, whereas in lower-grade astrocytomas they tend to be preserved.31
GLIOBLASTOMA
Glioblastoma multiforme (GBM) is the most malignant of the neoplasms with a primary astrocytic differentiation. Because it was thought to arise from the most primitive precursor of the stromal cell population (the glioblasts) and its gross morphology is highly variable and complex, it was given the name “glioblastoma multiforme.”33 The name is somewhat of a misnomer because GBMs are currently thought to arise from progressive dedifferentiation of mature astrocytes and not from rests of immature glioblasts. Glioblastomas are also referred to as grade IV astrocytoma.
Clinical Presentation
Surgical treatment of GBMs involves surgical debulking, which is typically followed by both radiation therapy and chemotherapy. Outcome, despite many treatment innovations, continues to be poor. The average survival from time of diagnosis varies significantly with patient age and tumor type (de novo vs. progression from previous low-grade astrocytomas) from 2 to 18 months. In patients younger than age 50, mean survival is less than 10 months and that drops to only 3 months in the eighth decade.34
Pathophysiology
GBMs are thought to arise from the progressive dedifferentiation of mature cells. They may arise from within a preexisting lower-grade astrocytoma or arise de novo. The genetics of GBMs that arise de novo are thought to differ from those that arise from a preexisting lower-grade glioma. Ironically, however, several authors continue to pursue bipotential precursor and neural stem cells or even the “brain tumor stem cell” as the culprits. A number of genetic abnormalities that predispose to the development of GBMs have been identified that may be acquired or inherited. Loss of tumor-suppressor genes on chromosomes 9, 10, 13, or 19 may lead to formation of a GBM. Allelic loss from chromosome 10 appears to be involved in up to 80% of GBMs.35
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


