CHAPTER 19 Intracranial Hemorrhage
Intracranial hemorrhage is defined as a pathologic distribution of hemorrhage within the calvaria. It is subdivided into extra-axial and intra-axial/intracerebral hemorrhage. Extra-axial hemorrhage includes blood in the epidural, subdural, and subarachnoid spaces. Intra-axial/intracerebral hemorrhage includes blood in the intraparenchymal and intraventricular spaces.
EPIDEMIOLOGY
Subarachnoid Hemorrhage
The worldwide incidence of nontraumatic subarachnoid hemorrhage (SAH) is estimated to be about 6 cases per 100,000 patient-years.1 SAH is seen predominantly in patients younger than 60 years of age. The principal causes of spontaneous SAH are ruptured aneurysms (85%), benign nonaneurysmal perimesencephalic (pretruncal) hemorrhage (10%), and rare causes (5%). Gender, race, and geographic region affect the incidence of SAH. Women have a 1.6 times greater risk of developing SAH than men. Black individuals have a 2.1 times greater risk than whites. The highest incidences of SAH occur in Japan (23 cases per 100,000 patient-years) and Finland (22 cases per 100,000 patient-years). The most important nonmodifiable risk factor for developing SAH is having a first-degree relative with SAH (increase in relative risk 6.6). Important modifiable risk factors are heavy alcohol consumption (relative risk 4.7), hypertension (relative risk 2.8), and smoking (relative risk 1.9).1
Intraparenchymal Hemorrhage
Intracranial hemorrhage is believed to occur in 10% to 30% of all strokes and is predominantly intraparenchymal in such cases. The exact incidence may be higher or lower, with more recent sources reporting a much lower incidence.2 Hypertension is the most common cause underlying most nontraumatic intracerebral hemorrhage (50%-60%). Hemorrhages associated with hypertension increase in frequency with age. Cerebral amyloid angiopathy (CAA) is the second most common cause of nontraumatic intraparenchymal hemorrhage and is responsible for 10% to 12% of all hemorrhagic strokes. As with hypertensive hemorrhage, the incidence of CAA-related hemorrhage increases with age. It is rarely encountered in patients younger than 70 years of age. Parenchymal hemorrhage may occur as a result of venous thrombosis, underlying neoplasm, vascular lesion, and inflammatory lesion. Hemorrhagic neoplasms include high-grade gliomas, vascular metastases, and extra-axial masses such as pituitary adenomas and (rarely) meningiomas. Underlying vascular lesions include arteriovenous malformations (AVMs), cavernous malformations, and, less commonly, telangiectatic AVMs, developmental venous anomalies (DVAs), and dural arteriovenous fistulas (DAVFs). Hemorrhagic inflammatory lesions include herpes simplex virus type 1 and angioinvasive fungal infections such as aspergillosis and mucormycosis.
CLINICAL PRESENTATION
Classically, nontraumatic SAH presents as an acute severe “thunderclap” headache, which patients may characterize as “the worst headache of their life.” Although only 1 in 10 patients presenting with thunderclap headache actually has SAH, headache remains the most common presenting complaint for SAH. It may be the sole presenting symptom in 25% to 33% of patients with SAH. The remainder of patients with acute severe headaches may suffer instead from symptoms of migraine or tension headache, viral meningitis, and so on. SAH also causes meningeal irritation that mimics meningitis, with meningismus, photophobia, prominent neck or upper back pain, seizures, and obtundation. Because the symptoms and signs of SAH are nonspecific, SAH is misdiagnosed clinically in 12% to 51% of cases.3 Failure to detect acute aneurysmal SAH has serious consequences, because (1) the rate of recurrent SAH from an untreated ruptured aneurysm is 50% in the first year and (2) recurrent hemorrhage predicts a poorer outcome.
PATHOPHYSIOLOGY
Nontraumatic, nonaneurysmal causes for SAH include arteriovenous malformations (AVM), infectious arteriopathies, and extension of intraparenchymal hemorrhage into the subarachnoid space. When SAH occurs in isolation anterior to the midbrain, the most likely cause is perimesencephalic (pretruncal) nonaneurysmal SAH. This entity most likely results from rupture of posterior fossa veins and occurs most commonly after sexual intercourse. It is regarded as benign, because recurrent hemorrhage is rare and the usual complications of SAH such as hydrocephalus and vasospasm are uncommon and/or mild.4
Intraparenchymal hemorrhage is most often the sequel of chronic systemic hypertension. Chronic arterial hypertension is believed to induce changes in small fragile vessels arising from the major proximal intracranial arteries. Fisher showed that chronic arterial hypertension causes histologic disorganization of the walls of small vessels and termed that pathology lipohyalinosis.5 Since then, lipohyalinosis has been understood to be either fibrinoid change or collagenous fibrosis, depending on which element predominates.6 Lipohyalinosis diminishes the integrity of the vessel wall, predisposes the patient to rupture of penetrating arterioles within the brain, and leads to hemorrhagic stroke. Hypertensive hemorrhage may also result from Charcot-Bouchard aneurysms, although these are less common than initially suspected. Hypertensive hemorrhages most commonly involve the basal ganglia and thalami of the deep supratentorial gray matter, the pons and midbrain of the brain stem, and the cerebellum. In common, all of these regions are supplied by small perforating arteries that arise from proximal intracranial arteries (e.g., proximal middle cerebral arteries and the basilar artery) and are therefore the sites most exposed to the deleterious effects of chronic systemic hypertension.
Cerebral amyloid angiopathy (CAA) is the second most important cause of intraparenchymal hemorrhage. It is clinically distinct from systemic amyloidosis and is caused by progressive deposition of β-amyloid protein in the media and adventitia of small to medium-sized vessels, resulting in the formation of twisted β-pleated sheet fibrils in the vessel walls.7 Histologic examination demonstrates staining with Congo red and deposition of crystals that appear birefringent under polarizing light microscopy. The abnormal protein weakens the structure of the vessel wall, causing fibrinoid necrosis, microaneurysms, and increased likelihood of hemorrhage. Recently, additional factors, such as the presence of apolipoprotein E4, have been shown to hasten the development of intraparenchymal hemorrhage in patients with CAA.8,9
Intraparenchymal hemorrhage can also result from reperfusion injury with acute hemorrhagic transformation of previously bland embolic arterial infarctions. After a clot occludes a vessel to cause infarction, naturally occurring fibrinolytic enzymes or thrombolytic therapy with tissue plasminogen activator (tPA) can cause lysis of the clot and restoration of blood flow to the damaged endothelial bed. The details underlying reperfusion damage have yet to be completely elucidated. However, a combination of oxidative radicals and metalloproteinases produced during reperfusion, and the thrombolytic agents themselves, can damage the endothelial cells, injure the basal lamina, and thereby open the blood-brain barrier, destroying the integrity of the cerebrovascular system.10,11 Increased vascular permeability then allows red blood cells to migrate into the surrounding extravascular space. Embolic infarctions typically undergo subacute mild hemorrhagic conversion. Revascularization of infarcted regions begins at 3 to 5 days after occlusion. Ingrowth of new immature vessels leads to extravasation of small amounts of blood into the infarcted brain.
The common causes for intraparenchymal hemorrhage are summarized in Table 19-1. Other less common causes of intraparenchymal hemorrhage include congenital bleeding diatheses (e.g., hemophilia, protein C and S deficiencies, von Willebrand factor deficiency), acquired coagulopathies (e.g., disseminated intravascular coagulopathy), and vasculitides.
TABLE 19-1 Common Causes of Nontraumatic Intraparenchymal Hemorrhage
IMAGING
CT
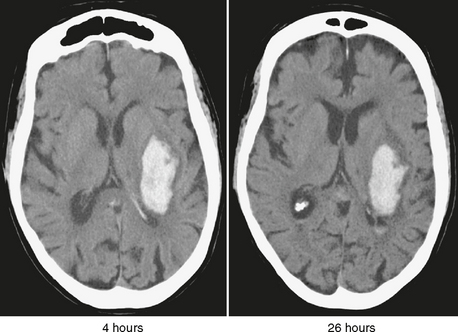
Acute hemorrhage can measure anywhere from 55 to 80 Hounsfield units (HU), higher than both normal gray matter (30-40 HU) and normal white matter (20-30 HU). In the clinical setting of severe anemia, acute hemorrhage can have a considerably lower attenuation and appear isodense to the brain. Although red blood cells begin to lose their integrity within hours of extravasation, complete cell lysis does not occur until later during the subacute stage of hemorrhage. In the first 12 hours after acute hemorrhage, the density of the blood often increases, because the hemoglobin becomes concentrated through clot formation and retraction. During the hyperacute period, the small amount of acellular serum expressed from the retracting clot forms a thin hypodense layer at the periphery of the hematoma (Fig. 19-1). Over the next 3 to 5 days, the brain surrounding the hemorrhage shows increasing lucency as proteases derived from the red cells induce vasogenic and cytotoxic edema and as an inflammatory response to the blood products disrupts the blood-brain barrier.