Introduction
Based on data collected from several central cancer registries during the years 2007-2011, the Central Brain Tumor Registry of the United States (CBTRUS) estimates that 67,900 new cases of primary brain and central nervous system (CNS) tumors will be reported in 2014—one third of which will be malignant. The brain and CNS are also common sites of secondary tumor implantation; metastases may occur up to 10 times more frequently than primary brain tumors.
The 2014 CBTRUS statistical report found that brain and CNS tumors are the most common neoplasm among those aged 0 to 19 years, with an average age-adjusted incidence rate slightly higher than leukemia. Children and young adults generally have better survival outcomes for most histologies; survival estimates have a large variation depending on tumor histology, ranging from a 5-year survival rate of 94% for pilocytic astrocytoma to 5% for glioblastoma.
The fourth edition of the World Health Organization (WHO) classification of tumors of the CNS was published in 2007 and continues to improve and expand histologic and clinical diagnostic criteria that enable the possibility of worldwide communication, clinical trials, and epidemiologic studies. In 2007 several new entities and histologic variants were added and genetic profiles were updated and discussed in a review article. The WHO-edited book publication includes concise commentary on the clinicopathologic characteristics of each tumor type. Box 9-1 is a list of tumors discussed in this chapter, which have been organized into clinically and imaging-relevant histology groups and reflect the 2007 WHO classification.
Gliomas
Astrocytic Tumors
Circumscribed
Pilocytic astrocytoma
Pleomorphic xanthoastrocytoma
Subependymal giant cell astrocytoma
Diffuse
Low-grade astrocytoma
Optic pathway glioma
Brainstem glioma
Anaplastic astrocytoma
Glioblastoma
Gliomatosis cerebri
Gliosarcoma
Oligodendrogliomas
Low-grade oligodendroglioma
Anaplastic oligodendroglioma
Ependymomas
Ependymoma
Anaplastic ependymoma
Subependymoma
Choroid Plexus Tumors
Choroid plexus papilloma
Choroid plexus carcinoma
Nonglial Tumors
Neuronal and Mixed Neuronal/Glial Tumors
Ganglioglioma
Gangliocytoma
Dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos)
Desmoplastic infantile ganglioglioma
Dysembryoplastic neuroepithelial tumor
Central neurocytoma
Pineal Parenchymal Tumors
Pineoblastoma
Pineocytoma
Embryonal Tumors
Medulloblastoma
CNS primitive neuroectodermal tumor
Tumors of the Cranial Nerves
Schwannoma
Neurofibroma
Malignant peripheral nerve sheath tumors
Tumors of the Meninges
Meningioma
Melanocytic tumors (melanocytoma, malignant melanoma, meningeal melanomatosis, diffuse melanocytosis)
Hemangioblastoma
Mesenchymal tumors
Lipoma
Chondrosarcoma
Rhabdomyosarcoma
Hemangiopericytoma
Lymphomas and Hematopoietic Neoplasms
Primary CNS lymphoma
Secondary CNS lymphoma
Granulocytic sarcoma (chloroma)
Germ Cell Tumors
Germinoma
Teratoma
Other germ cell tumors (embryonal carcinoma, yolk sac tumor, and choriocarcinoma)
Tumors of the Sellar Region
Pituitary adenoma
Microadenoma
Macroadenoma
Craniopharyngioma
Rathke’s cleft cyst
Tumors of the neurohypophysis (granular cell tumor, Langerhans cell histiocytosis)
Chordoma
Metastatic Tumors
Metastases to the brain
Metastases to the skull and intracranial dura
Leptomeningeal metastasis (leptomeningeal carcinomatosis)
Although many brain and CNS tumors are not histologically confirmed, particularly those with benign behavior, the most frequently reported histology overall in the United States from 2014 CBTRUS data was meningioma (36%) followed by glioblastoma (15%). Tumors of the pituitary gland and nerve sheath tumors combined account for about one fourth of all tumors, the vast majority of which are nonmalignant.
Clinical Presentation
The presenting symptoms in most patients with intracranial neoplasms are often nonspecific. Headache has been shown to occur in a majority of patients with primary brain tumors; indeed, headache is a common initial presentation for patients with high-grade gliomas. The headache is frequently described as diffuse and more severe in the morning and often dissipates in a few hours even without treatment. When unilateral, it can accurately indicate the hemisphere involved by tumor. Episodes of confusion or change in behavior also commonly bring the patient to the physician but can be confused with depression and result in delay of diagnosis. It is the persistent and progressive nature of the symptoms, often with development of visual disturbances or protracted nausea and vomiting, that eventually leads to consideration of brain tumor as a diagnostic possibility. Less common but more declarative presentations are focal neurologic symptoms (e.g., unilateral weakness, aphasia) and onset of seizures, the type and associated symptoms of which vary by tumor location. Seizures occur more frequently in low-grade gliomas and meningiomas than in higher-grade gliomas and lymphomas.
Intracranial neoplasms produce symptoms by three mechanisms: (1) infiltration and destruction of normal tissues (e.g., by a glioblastoma), (2) localized cortical irritation or depression (e.g., by a convexity meningioma), or (3) expansion within the rigid and unyielding cranial vault. Edema of the white matter adjacent to the tumor is often extensive and may account for much of the patient’s symptomatology and neurologic deficit, an impression that is verified by the striking improvement, both clinically and on imaging, exhibited by many patients after systemic administration of corticosteroids. An increase in intracranial pressure above the normal level of 180 mm H 2 O commonly accompanies an expanding intracranial mass. Headache, nausea, vomiting, and sixth cranial nerve palsy may reflect increased intracranial pressure. Prolonged or significantly elevated intracranial pressure manifests clinically as loss of attentiveness, apathy, and drowsiness. These signs of depressed cerebral function are most likely caused by diminished cerebral blood flow secondary to distention of the intracranial contents. This distention also produces traction on the overlying meninges, probably accounting for the headache, nausea, and vomiting that occur with chronic intracranial hypertension.
Diagnostic Imaging
Brain imaging is routinely performed using computed tomography (CT) or magnetic resonance imaging (MRI), typically after intravenous (IV) administration of iodinated contrast media (CT) or gadolinium contrast (MRI). The goals of diagnostic imaging in the patient with suspected intracranial tumor include detection of the presence of a neoplasm, localization of the extent of the tumor (including definition of involvement of key structures and assessment of the presence and severity of secondary changes [e.g., edema, herniation, hemorrhage]), and characterization of the nature of the process.
Imaging (usually MRI) is also extensively used in treatment planning before surgery or radiation to define the gross tumor margins and permit selection of the safest approach to the lesion. Increasingly, various functional/physiologic applications of MRI are employed preoperatively to determine the location of functionally eloquent cortex and critical white matter tracts, which can then be avoided during surgery. Interactive computerized neuronavigational devices use these data (1) to allow administration of very high doses of precisely focused radiation to small (up to 3 cm) intracranial tumors while sparing the adjacent normal brain and (2) to accurately register the previously acquired image data set onto intraoperative space, thus enabling precise intraoperative localization of surgical instrumentation relative to tumor margins, key vascular structures, and other critical anatomy. A continually evolving application is image-guided surgery, in which the entire surgical resection is guided by direct real-time MRI in the operating room environment.
Contrast Enhancement
IV administration of iodinated contrast medium causes a transient increase in the attenuation coefficient of normal gray matter and accentuates the difference in radiographic density on CT images between white matter and gray matter in the normal brain. Similarly, IV administration of paramagnetic contrast agent (e.g., gadolinium chelates) transiently induces shortening of both the T1 and T2 relaxation times of normal gray matter and accentuates the difference in MR signal intensity between normal cerebral gray matter and white matter. Most WHO grade IV and, to a slightly lesser extent, WHO grade III primary intracranial neoplasms and certain lower-grade primary brain tumors exhibit “contrast enhancement” in all or part of the tumor. The presence, extent, and pattern of tumor contrast enhancement have proved to be of significant value in improving detection, localization, and characterization of intracranial neoplasms by CT and MRI. Although the passage of intravascular contrast agent through brain parenchyma causes brief transient enhancement of the normal cerebral cortex, leakage of contrast material into the extravascular extracellular space accounts for the more pronounced and somewhat longer lasting contrast enhancement seen in many tumors and other structural abnormalities of the brain on both CT and MRI. The mechanisms involved in contrast enhancement of intracranial neoplasms on CT and MRI and in the uptake of radionuclide by such tumors appear to be identical.
In the normal brain, tight intercellular junctions between capillary endothelial cells create a blood-brain barrier that prevents escape of radionuclide or contrast agent from the intravascular space. In gliomas, electron microscopy studies have demonstrated that the endothelial junctions are frequently patent and the level of junctional patency is proportional to the degree of tumor malignancy. It therefore seems reasonable to postulate that the intensity of contrast enhancement in gliomas and other intracranial tumors reflects the degree of blood-brain barrier breakdown. Experimental data support this hypothesis by demonstrating that the peak intensity of tumor contrast enhancement in malignant gliomas occurs 20 to 60 minutes later than the peak serum concentration of contrast agent. Furthermore, glucocorticoids, which are known to diminish defects in the blood-brain barrier, also significantly reduce the extent of contrast enhancement in primary and secondary brain tumors.
Factors involved in maximizing contrast enhancement of intracranial neoplasms on CT or MRI include the quantity of injected contrast agent and the timing of the images. The standard dose of paramagnetic contrast agent (typically a chelate of gadolinium) administered IV for MRI is 0.1 millimoles per kilogram of body weight and is usually assessed on T1-weighted images. Application of a strong radiofrequency pulse that is slightly offset from the resonance frequency of water protons before initiation of the standard T1-weighted spin echo pulsing sequence produces saturation of protons in macromolecules, which is then transferred to the adjacent free water protons, thus diminishing their signal intensity. Because this maneuver, known as magnetization transfer (MT), does not affect the T1 shortening caused by IV administration of gadolinium, the intensity and conspicuity of resultant contrast enhancement are increased. However, because of greater suppression of white matter signal, MT alters tissue contrast relationships such that certain structures (basal ganglia, pulvinar, substantia nigra) become more conspicuous on noncontrast MT images, whereas other structures that normally enhance (choroid plexus, cerebral veins and dural venous sinuses, body of caudate nucleus, pituitary gland) do so more conspicuously on MT images acquired after contrast administration. The reader must therefore exercise caution in interpreting areas of apparent contrast enhancement as possibly signifying disease.
Complications of Brain Tumors
Brain Edema
Edema or swelling of the brain is a common accompaniment of many brain tumors and other structural abnormalities of the brain. When sufficiently severe, edema may be responsible for both focal and generalized signs of brain dysfunction. Edema is not a single pathologic response to a variety of insults but rather occurs in at least three different forms: vasogenic (secondary to tumor, inflammation, hemorrhage, extensive infarction, or contusion), cytotoxic (in response to hypoxia, early infarction, or water intoxication), and interstitial (resulting from acute obstruction to the flow or absorption of cerebrospinal fluid [CSF]).
Vasogenic edema is the form of brain swelling most typically associated with intracranial neoplasms. It is caused by a breakdown in the blood-brain barrier with seepage of a plasma filtrate containing plasma proteins through patent junctions between capillary endothelial cells. Gray matter is relatively resistant to the development of edema, and the extracellular plasma filtrate mainly accumulates in the white matter, extending along the major white matter fiber tracts. The subcortical arcuate (U) fibers between adjacent gyri offer greater resistance to the spread of edema than the long white matter tracts and are therefore involved relatively later and in more severe cases.
The severity of edema associated with various brain tumors varies widely even between lesions of identical histology. In general, white matter swelling is greatest in association with carcinomatous metastases, and it is not unusual for a small metastasis to provoke a disproportionately large amount of edema. In order of declining severity, edema is associated with metastases, glioblastomas, meningiomas, and low-grade gliomas, but exceptions to this order are common. Rarely a low-grade glioma may be surrounded by extensive edema, whereas occasional metastases or meningiomas may have little associated white matter swelling.
On CT, vasogenic edema appears as widening and diminished density of the major white matter tracts with fingerlike extensions into the arcuate fibers in each gyrus. The overlying cortical gray matter is compressed and thinned by the expanded pseudopods of edematous white matter (see Figs. 9-14 and 9-17 ).
On noncontrast MRI, the high water content in the edematous peritumoral white matter causes prolonged T1 and T2 relaxation in the involved white matter; these findings manifest as an increase in signal intensity on T2-weighted images and as a less conspicuous decrease in signal intensity on T1-weighted images (see Figs. 9-1A, 9-15, and 9-20B ). It is often difficult to delineate the boundary between tumor and edema on these noncontrast images; IV administration of paramagnetic contrast agent permits gross demarcation on T1-weighted (see Fig. 9-20C ) and fluid-attenuated inversion recovery (FLAIR) images in many tumors (e.g., metastases) but not in malignant gliomas.
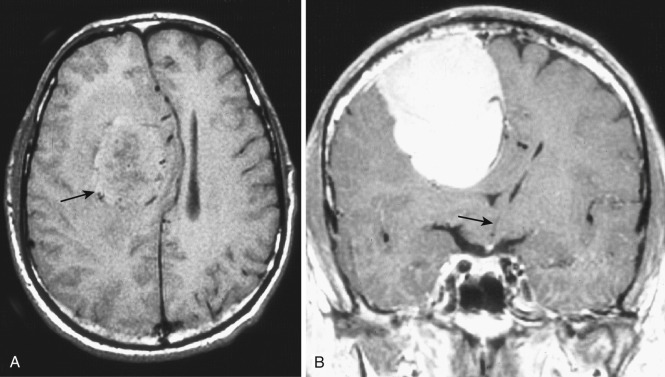
Systemic administration of glucocorticoids minimizes the blood-brain barrier defect inherent in most higher-grade brain tumors, with resultant reduction in fluid and protein extravasation. Diminution in peritumoral white matter swelling as well as in the volume and intensity of tumor contrast enhancement is often evident on follow-up imaging studies within a few days after institution of steroid therapy.
Interstitial edema secondary to obstruction of CSF pathways appears on CT as poorly circumscribed periventricular hypodensity and on T2-weighted MRI as bandlike periventricular hyperintensity of varying thickness and margination. These findings are often symmetric and are most evident surrounding the anterosuperior margins of the dilated frontal horns of the lateral ventricles (see Fig. 9-2D ) and the posterior margins of the occipital horns. Fluid accumulates in the periventricular white matter as a result of transependymal seepage of ventricular fluid across microscopic breaks in the ependymal lining of the ventricles. Systemic glucocorticoids do not affect this type of edema, but surgical insertion of a ventriculostomy shunt catheter above the level of obstruction usually results in prompt decompression of the ventricles and disappearance of the periventricular hypodensity/hyperintensity.
Herniations
An expanding mass within the rigid cranial vault, whether due to tumor, edema, or a combination of both processes, compresses and distorts the adjacent normal brain, producing internal herniation of the brain under the relatively rigid falx or through the tentorial incisura.
Laterally placed masses in the cerebral hemispheres, particularly those located in the superior temporal, midfrontal, or frontoparietal regions, displace the deep central structures (basal ganglia, thalamus, third ventricle, lateral ventricles, septum pellucidum) medially. The ipsilateral cingulate gyrus is compressed and displaced across the midline under the free edge of the falx (subfalcine herniation). Medially located high frontoparietal (parasagittal) masses depress and displace the cingulate gyrus contralaterally but without significantly affecting the deep central structures. Subfalcine herniation is most clearly depicted on coronal images, which also demonstrate depression and contralateral displacement of the corpus callosum and the underlying body of the ipsilateral ventricle ( Fig. 9-1 ; see Fig. 9-11 ). The septum pellucidum and third ventricle are bowed away from the side of the mass. On axial CTs and MRIs, the ipsilateral ventricle appears compressed and displaced contralaterally.
Tumors of the temporal lobe and middle cranial fossa displace the uncus and parahippocampal gyrus on the medial aspect of the temporal lobe toward the midline, with resultant encroachment on the lateral aspect of the suprasellar and ambient (circummesencephalic) cisterns and the tentorial incisura (descending transtentorial herniation) (see Figs. 9-7 and 9-18A ). The ipsilateral margin of the midbrain is compressed, displaced contralaterally, and rotated by the impinging temporal lobe. Noncontrast MRI, postcontrast CT, or both may demonstrate medial displacement of the posterior communicating and posterior cerebral arteries, narrowing of the contralateral crural and circummesencephalic cisterns, and widening of their ipsilateral counterparts behind the impinging temporal lobe. Late secondary signs include hemorrhages in the compressed midbrain and unilateral or bilateral medial occipital infarctions (due to occlusion of one or both posterior cerebral arteries). Large centrally located cerebral masses for which the primary vector of force is directly downward may cause bilateral transtentorial herniation.
Ascending transtentorial herniation with upward displacement of the superior cerebellar vermis and hemisphere through the posterior aspect of the tentorial incisura is caused by expanding masses in the superior portion of the posterior compartment of the posterior cranial fossa, including the upper half of the cerebellum. As the superior cerebellar vermis is forced anteriorly and superiorly into the incisura, it protrudes into and compresses the superior cerebellar cistern from below and flattens the normal posterior convexity of the quadrigeminal cistern from behind. Increasing severity of herniation leads to reversal of that convexity and eventually to obliteration of the quadrigeminal and superior cerebellar cisterns ( Fig. 9-2A and B ) with flattening of the posterior margin of the third ventricle.
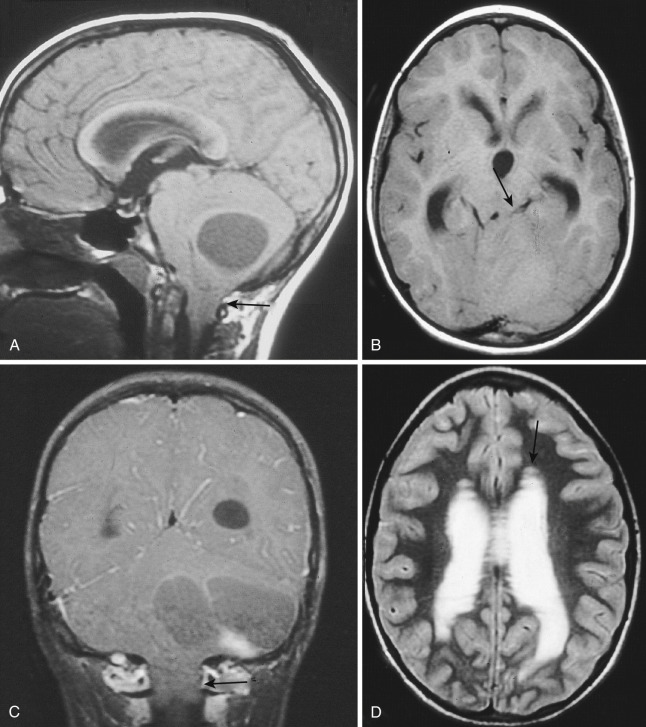
Tumors of the lower half of the cerebellum and other masses in the inferior portion of the posterior compartment of the posterior fossa may produce downward displacement of the cerebellar tonsils through the foramen magnum (tonsillar herniation). This situation can be visualized directly on T1-weighted MRI in the sagittal and coronal planes (see Fig. 9-2A and C ) and can be suggested on axial CT or MRI if the cisterna magna and upper cervical posterior subarachnoid spaces are encroached on and partially or completely obliterated by soft tissue density.
Hemorrhage
Hemorrhage is not a common accompaniment of most intracranial neoplasms at initial presentation. In a series of 973 intracranial tumors, CT demonstrated acute intratumoral bleeding in 28 patients (3%) at the time of initial diagnosis and in 7 additional patients (0.7%) who experienced clinical deterioration during their subsequent course. In Cushing and Eisenhardt’s meticulously reported experience, hemorrhage was found in association with intracranial gliomas in 31 of 832 cases (3.7%) (see Fig. 9-14 ). Acute hemorrhage was demonstrated on CT in 6 of a series of 131 meningiomas (4.6%).
Intratumoral bleeding is more common in certain tumors, notably metastases from choriocarcinoma, melanoma (see Fig. 9-63 ), carcinomas of the lung and thyroid, and renal cell carcinoma, as well as in neuroblastoma, lymphoma, and medulloblastoma. In metastatic melanoma and choriocarcinoma, the typically hyperdense appearance of the metastatic nodules on CT has been attributed to the presence of acute hemorrhage or hemosiderin within the tumor.
Because many of the breakdown products of blood have paramagnetic properties, MRI provides a unique and highly sensitive method for detection of subacute or chronic intracranial bleeding. Hemorrhage associated with intracranial tumors may occur centrally within a necrotic tumor cavity (in glioblastoma [see Fig. 9-14 ] and some metastases) or peripherally around the tumor (seen in other metastases and meningiomas). Signal intensity (MRI) or density (CT) is typically more heterogeneous in intratumoral hemorrhage than in benign intracranial hemorrhage. On occasion, hemorrhage may completely mask the presence of the underlying neoplasm, but images obtained after injection of contrast medium may demonstrate contrast enhancement of tumor along a margin of the hematoma or in other locations within the brain. Hemorrhage into a pituitary adenoma is a common cause of “pituitary apoplexy” and may obliterate not only the entire tumor but also the normal pituitary (see Figs. 9-56 and 9-57 ).
Types of Brain Tumors
Gliomas
Gliomas include all primary brain tumors of astrocytic, oligodendroglial, or ependymal origin—astrocytomas, oligodendrogliomas, and ependymomas—as well as choroid plexus papillomas and carcinomas.
Astrocytomas.
Astrocytomas (tumors derived from astrocytes) are the most common of all primary intracranial neoplasms. In the 1993 revision of the WHO classification of brain neoplasms, astrocytomas were subdivided into four histologic grades. This histologic grading system has demonstrated a high positive correlation with the biological potential and behavior of tumors. Tumors of lower histologic grades (I and II) demonstrate few mitoses, little cellular structural variation (pleomorphism), and no vascular proliferation or necrosis. Tumors in grades III (anaplastic astrocytoma) and IV (glioblastoma multiforme) are characterized by more frequent mitoses, higher degrees of cellular dedifferentiation, and increasing angioneogenesis at the tumor periphery and by necrosis within the more central portion of the tumor.
Astrocytomas are characterized on both gross dissection and diagnostic imaging into two groups: circumscribed and diffuse. In general, the diffuse astrocytomas are more common, tend to occur more in adulthood, and are more infiltrative or aggressive with spread along the white matter tracts.
Circumscribed astrocytomas.
Three histologic types of astrocytoma are assigned to the circumscribed group. They are pilocytic astrocytoma (PA), pleomorphic xanthoastrocytoma (PXA), and subependymal giant cell astrocytoma (SCGA). All three types occur mainly in younger individuals, are more localized, and have a less aggressive biological potential than the diffuse astrocytomas.
Pilocytic astrocytoma.
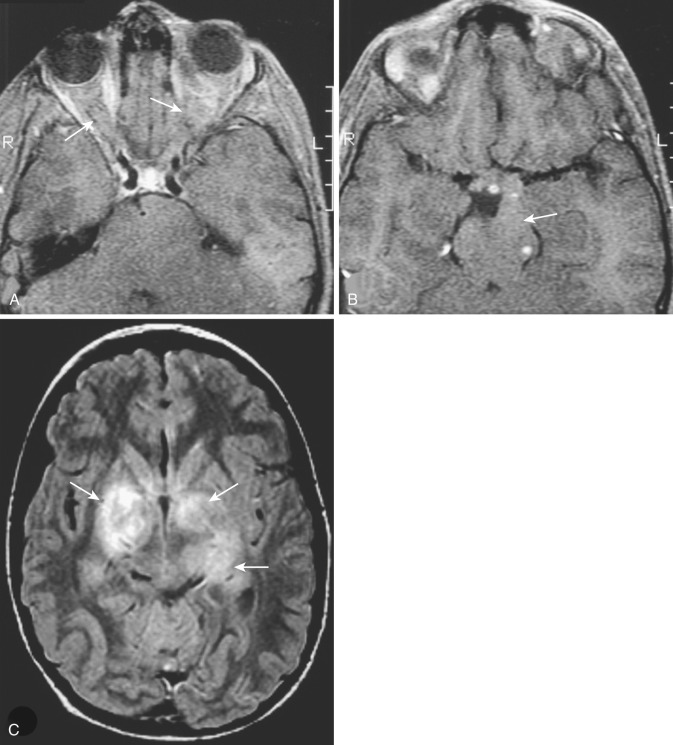
The most common type of circumscribed astrocytoma, PA, occurs in the cerebellum (60% of cases) in children ( Fig. 9-3 ; see Fig. 9-2 ), with a peak age distribution between 5 and 15 years. PA also frequently (30% of cases) originates in the optic pathways and the adjacent hypothalami ( Fig. 9-4 ), usually in slightly younger children (2-3 years), often in association with neurofibromatosis type 1 (NF1).
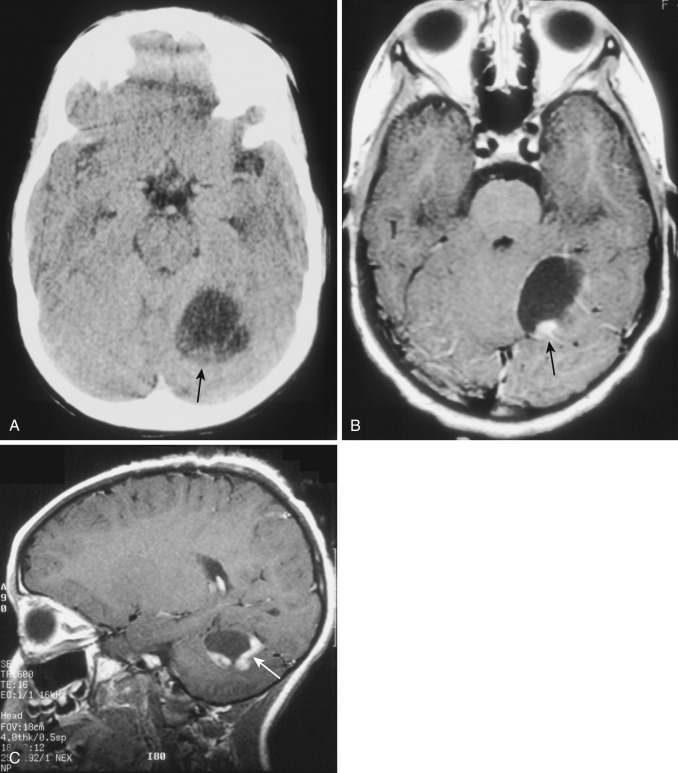
PA is the classic well-circumscribed brain neoplasm, but it lacks a true tumor capsule. It grows mainly by expansion rather than by infiltration. It is widely considered to be a benign (biologically stable) neoplasm and is classified histologically as grade I. It may be densely cellular or more loosely arranged with intervening microcysts, and both patterns may coexist in different portions of the same tumor. Histologic characteristics include the frequent presence of eosinophilic Rosenthal fibers in tumor cell cytoplasm, although these can also be seen in reactive gliosis. Mitoses are rare in this lesion, and necrosis is not seen. Most often the tumor represents a mural nodule in the wall of a well-circumscribed cyst. The origin and nature of the cyst are not well understood, but the cyst fluid is proteinaceous and likely secreted from coiled capillaries found in the midst of the nodular tumor. With the exception of the mural tumor nodule, the wall of the cyst consists of compacted normal brain or nonneoplastic gliotic tissue. Intratumoral calcification is occasionally identified (5%-25% of cases), but hemorrhage into or adjacent to the tumor is rare.
The clinical presentation of children with PA depends on the tumor site. The cerebellar tumors typically manifest with headache, nausea, and vomiting because of hydrocephalus secondary to obstruction of the fourth ventricle. Weakness and loss of equilibrium are not uncommon. The optic tumors typically cause difficulties with vision, but signs of hydrocephalus may appear if the tumor has extended into the hypothalamus and is obstructing the third ventricle.
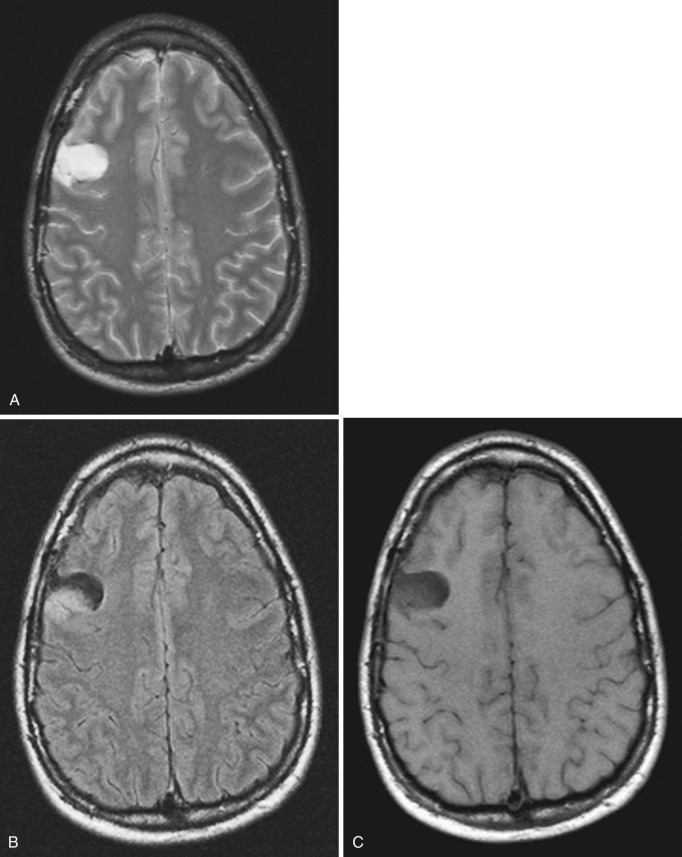
On CT the typical cerebellar PA appears as a smoothly marginated hypodense cystlike mass with a well-defined less hypodense tumor nodule on one wall (see Fig. 9-3A ). The nodule may contain one or more areas of dense calcification. The cyst fluid is less hypodense than the CSF (15-25 Hounsfield units [HU]) because of its high protein concentration. Typically after IV administration of a contrast agent, dense homogeneous contrast enhancement of the mural nodule but not of the other walls of the cyst is seen (see Fig. 9-3B and C ). More extensive enhancement of the cyst walls suggests a more aggressive (higher histologic grade) tumor.
On MRI the mural nodule appears homogeneously hyperintense to gray matter on T2-weighted images and hypointense to isointense on T1-weighted images. Intratumoral calcification may cause a more heterogeneous appearance. The associated cyst is even more hyperintense on T2-weighted images and even more hypointense on T1-weighted images (see Fig. 9-2A ). Edema of the adjacent white matter is usually minimal. Homogeneous contrast enhancement of the tumor nodule is characteristic (see Figs. 9-2C and 9-3B and C ), although a calcific focus (if present) does not demonstrate enhancement.
The clinical prognosis of cerebellar PA is guardedly optimistic. The cerebellar tumor nodule can often be totally excised, and long-term survival rates exceeding 75% are commonly reported. The location and extent of the optic-chiasmatic-hypothalamic tumors typically preclude total excision, but irradiation and chemotherapy often achieve long-term tumor control.
Optic pathway PA manifests on both CT and MRI with enlargement of the optic nerve as an intraconal mass, with characteristic kinking or buckling of the nerve secondary to the neoplasm itself and vascular congestion. Isointensity on T1-weighted MRI and heterogeneous hyperintensity on T2-weighted MRI are typical, with variable enhancement following IV contrast material administration.
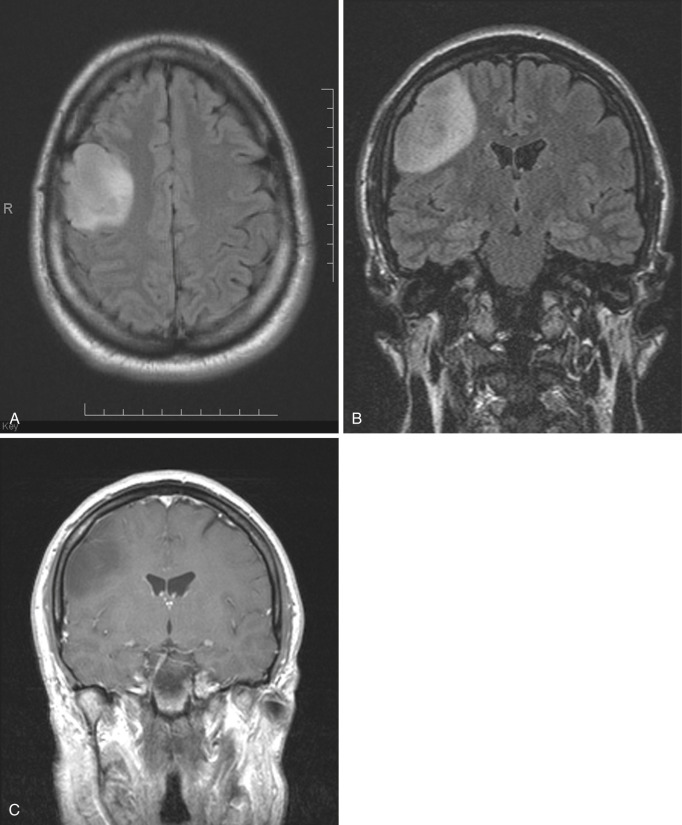
Pleomorphic xanthoastrocytoma.
PXA was newly designated in the 1993 WHO revised classification. The number of cases documented in the literature is still small. PXA is presumed to arise from subpial astrocytes near the surface of the cerebral hemispheres. Rare tumors have also been described in the posterior cranial fossa and in the spinal cord. The gross morphology resembles the mural nodule and cystic appearance of cerebellar PA. The nodule is often based on the pial surface of the brain.
On CT and on MRI the appearance is similar to that of PA but in a different location: well circumscribed, hypodense to isodense on CT, hypointense on T1- and hyperintense on T2-weighted MRI, often with a cystic component that is isointense to CSF ( Fig. 9-5A ). However, the location is supratentorial (in order of descending frequency: temporal, frontal, parietal) and superficial, with little or no mass effect. Although the tumor presents a circumscribed appearance, it usually grows slowly and may infiltrate the adjacent brain and the overlying pia, even causing focal thickening of the meninges (dural tail). Intense homogeneous contrast enhancement of the solid portion of the tumor and the overlying meninges is the rule (see Fig. 9-5B and C ).
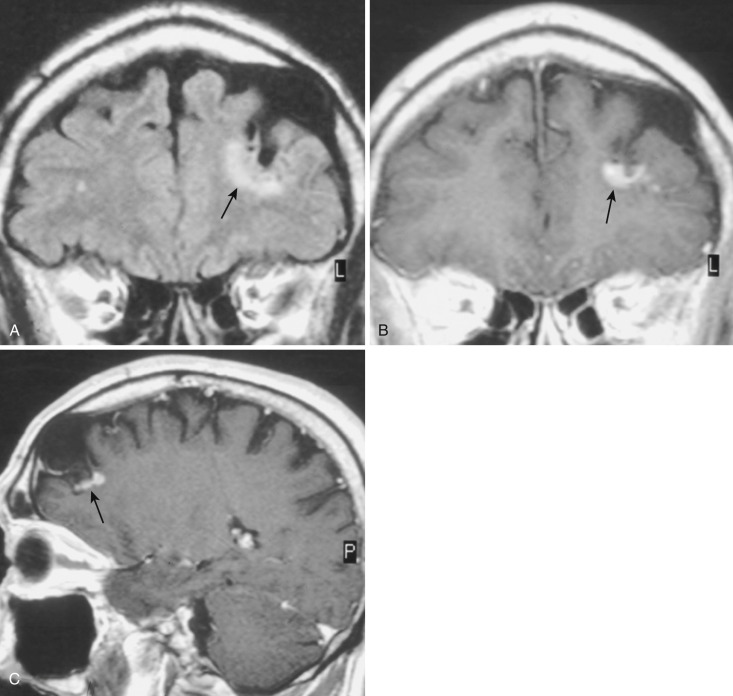
Histologically, as the name implies, the tumor nodule is pleomorphic, but cells with a foamy myxoid cytoplasm are common. PXA is usually classified as grade II, but later dedifferentiation into higher-grade neoplasm may occur. The clinical presentation most commonly consists of seizures that may be intractable. Most reported cases have been in adolescents or young adults; the median age at diagnosis is 26 years. Treatment is usually surgical. Malignant transformation has been described in 10% to 25% of cases, but the 10-year survival is reported as 70%.
Subependymal giant cell astrocytoma.
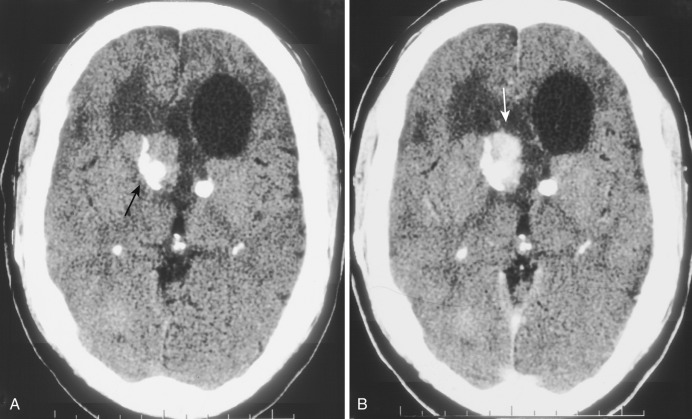
This low-grade (WHO grade I) tumor occurs almost exclusively in patients with tuberous sclerosis. It is estimated that 10% to 15% of patients with tuberous sclerosis develop this neoplasm, usually in the late first or second decade of life. These tumors arise from subependymal nodules located on the lateral wall of the lateral ventricle overlying the head of the caudate nucleus. By convention, subependymal nodules that are more than 1 cm in diameter or become symptomatic are considered giant cell astrocytomas. The tumor does not invade into the underlying caudate head but rather grows exophytically into the ventricular lumen near the foramen of Monro, which may be obstructed either unilaterally or bilaterally. Despite this tendency for intraventricular growth, the overlying ependyma remains intact and dissemination via CSF is rare. Intratumoral calcification is common and may be heavy.
Histologically the tumor is seen to contain large multinucleated giant cells of uncertain origin. On cytochemical analysis, some of these cells display GFAP (glial fibrillary acidic protein) reactivity, a feature consistent with an astrocytic origin, whereas others contain neuron-specific enolase, a finding consistent with neuronal origin.
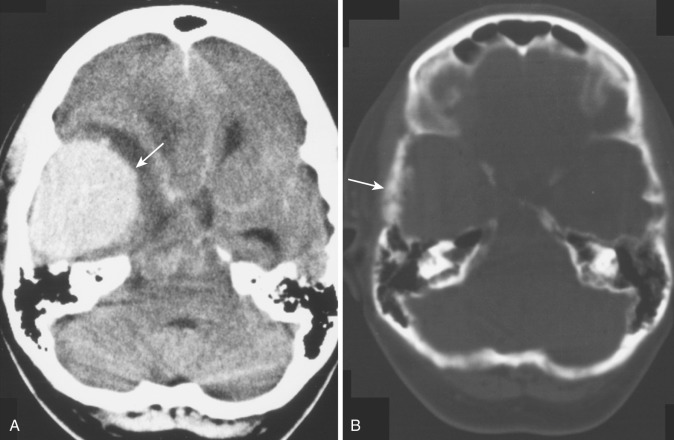
Subependymal giant cell astrocytomas present a characteristic appearance on CT. They appear as large, hypodense, polypoid, sharply marginated intraventricular masses at the level of the foramen of Monro ( Fig. 9-6A ). The tumor may be heavily calcified and may obstruct the foramen unilaterally or bilaterally, causing gross enlargement of one or both lateral ventricles. Other manifestations of tuberous sclerosis are usually evident, including cortical tubers and subependymal hamartomatous nodules. On MRI, subependymal giant cell astrocytoma appears as a heterogeneous sharply demarcated intraventricular mass that is mildly hyperintense on T2-weighted images and hypointense or isointense on T1-weighted images. It is located within the frontal horn of the left ventricle, without evidence for deep invasion or spread within the basal ganglia. On both CT and MRI, intense but heterogeneous contrast enhancement is often demonstrated (see Fig. 9-6B ). The heterogeneous signal on MRI both without and with contrast enhancement reflects the presence of dense calcification in these tumors. Differentiation of this tumor from the subependymal hamartomatous nodules that are so ubiquitous in patients with tuberous sclerosis is sometimes problematic, but the factors usually considered are the characteristic tumor location, its larger size, and the presence of contrast enhancement; contrast enhancement is not seen in the nontumorous nodules. Clinically these patients present with signs of increased intracranial pressure (headache, nausea, vomiting, and obtundation) due to obstruction of the foramen of Monro. Treatment is surgical, and the recurrence rate is low if the resection is complete.
Diffuse astrocytomas.
Poorly marginated diffuse astrocytomas demonstrate much more aggressive infiltrative behavior than circumscribed astrocytomas. Progression from lower histologic grade (astrocytoma, WHO grade II) to higher grades (anaplastic astrocytoma, WHO grade III; glioblastoma, WHO grade IV) is common; it is estimated that 50% of low-grade astrocytomas undergo subsequent dedifferentiation into higher-grade tumors (anaplastic astrocytoma or glioblastoma).
Diffuse astrocytomas are characterized genetically by inactivation of the TP53 gene, which is located on the short arm of chromosome 17. The TP53 gene encodes a protein that is involved in regulation of the cell cycle and DNA formation. Inactivation of this gene by mutation may lead to formation of abnormal DNA as well as other changes that can contribute to malignant transformation of the cells. It is interesting to note that primary glioblastomas that arise de novo and not from preexisting lower-grade astrocytoma do not exhibit loss of the TP53 gene.
Low-grade astrocytoma.
Low-grade (WHO grade II) supratentorial astrocytomas are hypercellular tumors that consist of well-differentiated fibrillary or gemistocytic astrocytes and demonstrate few mitoses and moderate pleomorphism but no vascular proliferation or necrosis. On immunohistochemistry studies, they demonstrate strong affinity for GFAP. They arise in white matter and grow by infiltration predominantly along white matter tracts, extending between and separating anatomic landmarks. The involved area of the brain eventually appears larger than its contralateral counterpart. Cerebral astrocytomas are usually solid masses that, because of their infiltrative characteristics, are poorly circumscribed and blend imperceptibly with the adjacent cerebral parenchyma. Small and large cysts are occasionally found within these tumors, but hemorrhage is rare.
Approximately two thirds of low-grade astrocytomas are supratentorial; these arise mainly in the frontal, parietal, and temporal lobes ( Figs. 9-7 and 9-8 ). They typically provoke little or no edema in the surrounding tissues. Because such a tumor begins in the white matter without affecting the more eloquent cortical centers, symptoms are usually vague and nonlocalizing until the lesion gains sufficient size, extends into eloquent cortex, or undergoes dedifferentiation into a grade III or IV tumor. The peak age range for occurrence of low-grade astrocytoma is the third through fifth decades of life.
Some diffuse (initially low-grade) astrocytomas occur in childhood, notably involving the optic nerves and chiasm, optic tracts, and visual pathways as well as the hypothalamus and thalamus. More than half of the optic pathway gliomas of early childhood involve the optic chiasm and hypothalamus, and 40% of these are diffuse fibrillary astrocytomas. In contradistinction to the young child with NF1 in whom more circumscribed PAs develop along the visual pathways, the more diffuse fibrillary astrocytomas arise without any identifiable genetic predilection, although in a similar age group (peak age, 2-3 years); these are termed sporadic optic pathway astrocytomas. However, the diffuse fibrillary tumors share with their adult counterparts a poorer prognosis and thus merit a more aggressive course of management.
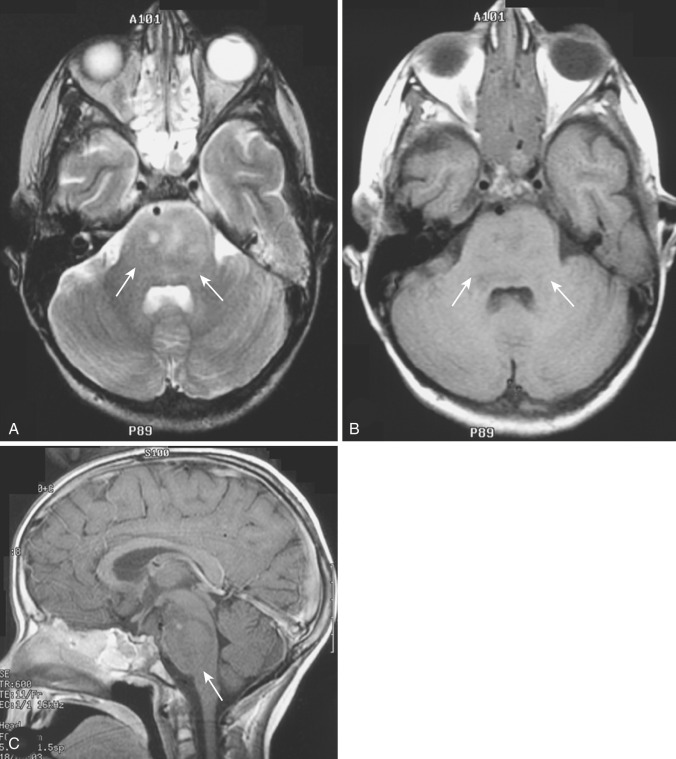
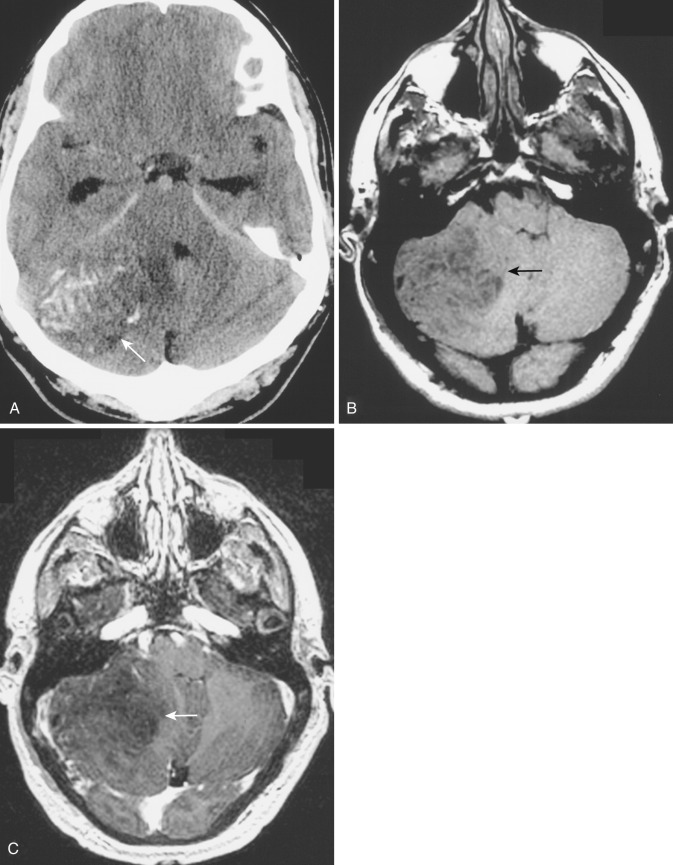
Another relatively common location of diffuse low-grade astrocytoma in childhood is in the brainstem. Brainstem gliomas constitute 10% to 20% of all pediatric brain tumors. The peak age of occurrence is 5 to 10 years. These tumors arise mainly in the pons but commonly involve the midbrain and medulla and may extend into the upper cervical spinal cord. Despite their proximity to the aqueduct and fourth ventricle, considerable tumor growth may take place without causing obstruction of the ventricular system. Multiple cranial nerve deficits, long tract signs, and ataxia are the most common presenting findings. Although these tumors are often regarded as low-grade lesions, histologic heterogeneity with intermixed areas of higher-grade neoplastic change is common. Approximately 60% of brainstem gliomas demonstrate foci of glioblastomatous change with central necrosis. Five-year survival rates range from 15% to 30%.
On CT, low-grade diffuse cerebral astrocytomas appear most commonly as poorly marginated areas of decreased attenuation involving the white matter of one or more lobes of the brain with mild to moderate mass effect. A more heterogeneous appearance with patchy areas of normal or slightly higher tissue density intermixed with hypodensity is seen in a small number of cases. Contrast enhancement is usually absent, reflecting a lack of tumor vascularity, blood-brain barrier breakdown, or both.
Because low-grade astrocytomas are predominantly hypodense (15-30 HU) and usually do not exhibit contrast enhancement, it is often not possible with CT to differentiate the margin of the tumor from the adjacent white matter. However, because the blood-brain barrier is intact, extensive peritumoral edema is unusual in low-grade astrocytomas. Grossly identifiable areas of intratumoral hemorrhage or necrosis are also not typically seen on CT or MRI studies in these lesions. Calcification is identified in 20% of low-grade diffuse astrocytomas, and cysts are occasionally demonstrated. The presence of hemorrhage, necrosis, edema, and/or contrast enhancement suggests progression to a higher tumor grade.
Optic pathway diffuse astrocytomas appear on CT and MR as rounded or lobulated thickening of the more posterior optic pathway, typically involving the optic chiasm and retrochiasmal segment of the optic pathway. This typically appears homogeneously hypodense on CT, T1 isointense to brain, and variably hyperintense on T2-weighted images. The tumor may encase the arteries of the circle of Willis and invade the floor of the third ventricle, causing obstructive hydrocephalus. Necrosis and hemorrhage are uncommon. In contrast to optic pathway PAs, cystic foci may be seen. Contrast enhancement is highly variable and typically does not signify malignant progression, a rare occurrence in sporadic diffuse optic pathway astrocytoma in childhood.
Brainstem glioma typically appears on CT as ill-defined diffuse heterogeneously hypodense and isodense asymmetric expansion of the pons and medulla, with dorsal displacement of the floor of the fourth ventricle and aqueduct and ventral encroachment on the pontine cistern ( Fig. 9-9A and B ). Despite the impression of the tumor on the aqueduct and the floor of the fourth ventricle, hydrocephalus is a relatively late finding. Approximately 50% of brainstem gliomas exhibit varying degrees of contrast enhancement on CT and MRI after IV administration of contrast medium (see Fig. 9-9C ). Contrast enhancement at presentation suggests higher grade and is usually patchy and heterogeneous. However, new enhancement after treatment does not necessarily suggest disease progression but may actually represent treatment response.
On MRI, diffuse low-grade astrocytomas appear relatively homogeneous and demonstrate varying degrees of prolonged T1 and T2 relaxation. They therefore appear homogeneously hypointense on T1-weighted images and hyperintense on T2-weighted images in comparison with normal brain (see Figs. 9-7A and 9-8A and B ). Because of the inherently higher contrast resolution of MRI, astrocytomas are often better demarcated from adjacent normal white matter on MRI than on CT, reflecting their higher water content. Nevertheless, as on CT, the apparent margins of the lesion on MRI (without or with contrast enhancement) do not closely correlate with histopathologic evidence of tumor extent. As on CT, there is no grossly evident contrast enhancement on MRI in diffuse low-grade cerebral astrocytomas (see Figs. 9-7B and 9-8C ); the demonstration of contrast enhancement should raise the strong suspicion of a higher tumor grade.
The differential imaging diagnosis of low-grade diffuse astrocytoma on CT and MRI is ubiquitous. Principal considerations are cerebral infarction, cerebritis, and higher-grade glioma (e.g., anaplastic astrocytoma). Infarctions tend to involve overlying cortical gray matter more extensively and are often more sharply delineated from the adjacent cerebral parenchyma. Inflammatory processes and higher-grade gliomas are often associated with blood-brain barrier breakdown and exhibit contrast enhancement and surrounding edema.
The prognosis in low-grade astrocytoma is guarded. Many of these tumors in young adults progress to a higher grade. Median survival has been reported as 5 to 8 years.
Anaplastic astrocytoma.
As the name implies, anaplastic astrocytomas (WHO grade III) exhibit considerable variation in cellular morphology, with high cellularity, frequent mitoses, and foci of vascular proliferation, findings that are not present in low-grade (grade II) astrocytomas. However, no necrosis is present in anaplastic astrocytomas on either gross inspection or microscopic evaluation. These tumors tend to occur in a slightly older age group (peak age, mid-40s) than the grade II lesions and often represent a progressive dedifferentiation of a previously low-grade tumor.
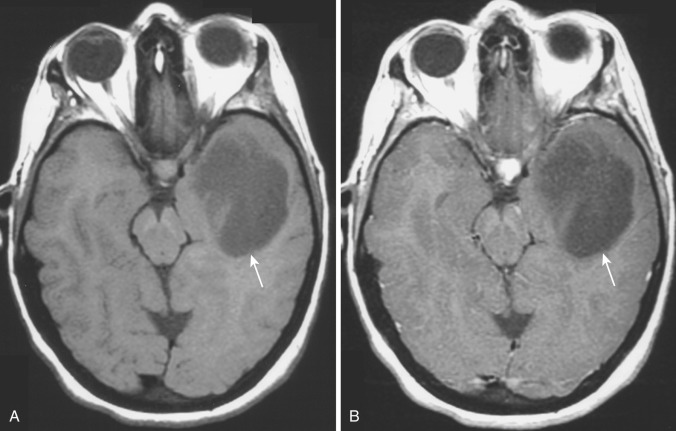
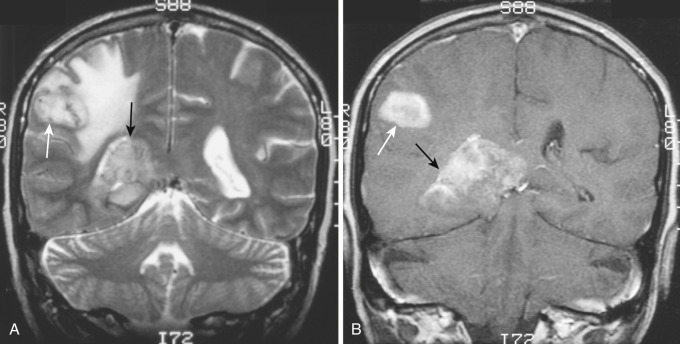
Reflecting the increased histologic variation, the imaging findings are also less homogeneous than in the lower-grade tumors ( Figs. 9-10A and 9-11A ). On T2-weighted MRI, anaplastic astrocytomas are most often heterogeneously hyperintense, frequently involve the overlying cortex, and are associated with a greater degree of vasogenic edema and overall mass effect (see Fig. 9-11A ). Restricted diffusion is often identified within these tumors, likely reflecting foci of increased cellularity. Contrast enhancement is common on both CT and T1-weighted MRI (see Figs. 9-10B and 9-11B and C ); compared with glioblastoma (grade IV) the enhancement pattern is often not ringlike but rather patchier. Although the region of contrast enhancement may appear discrete, infiltration of tumor cells beyond the margin of enhancement into the edematous surrounding white matter is nearly always present, often referred to as infiltrative edema.
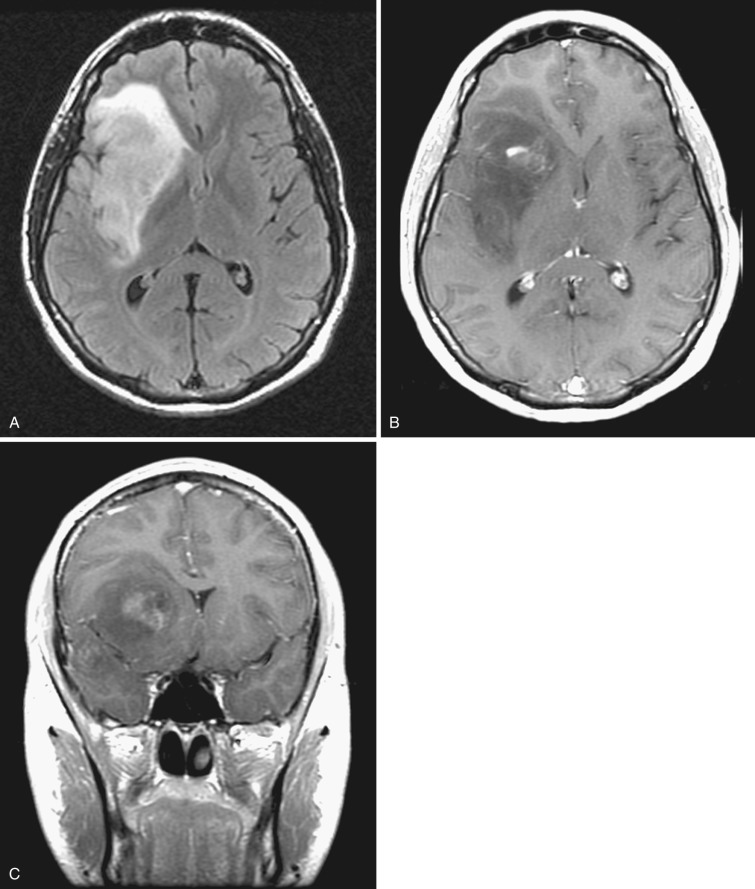
Current estimates indicate that anaplastic astrocytomas represent about 25% of all primary gliomas of the brain. Median survival for patients with anaplastic astrocytoma is 2 to 3 years. Most tumors progress relatively rapidly to frank glioblastoma with intratumoral necrosis.
Glioblastoma.
Glioblastoma (WHO grade IV) is the most common primary malignant tumor of the CNS, accounting for more than half of all intracranial gliomas. It is biologically the most aggressive of the gliomas, with rapid progression of clinical signs and symptoms and a mean survival time in the range of 12 months. Like all other astrocytomas, glioblastoma characteristically involves the cerebral white matter, but this highly malignant process readily infiltrates and destroys the adjacent gray matter with loss of gray-white differentiation. As their name implies, these highly malignant tumors have a variegated histologic appearance with interspersed areas of hypercellularity, cellular pleomorphism, endothelial proliferation, and intratumoral necrosis.
It is probable that most glioblastomas arise by anaplastic change within preexisting low-grade cerebral astrocytomas, although some likely occur de novo. Although most commonly focal and singular in their presentation, glioblastomas may be multifocal or multicentric in origin. The peak age of incidence of glioblastoma is during the fifth and sixth decades of life, some 10 to 20 years later than the peak age for astrocytoma. However, glioblastoma is not rare in younger adults or even in children; of all glioblastomas, approximately 3% occur in childhood, mainly in the brainstem and cerebellum.
On gross inspection, glioblastomas appear more or less spherical when confined to one lobe of a cerebral hemisphere. However, these rapidly growing neoplasms invariably infiltrate into adjacent lobes, the corpus callosum (“butterfly gliomas”), and the contralateral hemisphere. Tumors that have extended widely tend to have more irregular configurations with lobulated margins of varying thickness. Infiltration of the ependyma of the lateral and third ventricles and penetration of the cerebral cortex with invasion of the overlying meninges occur commonly and may lead to seeding through the CSF pathways. Vascular endothelial proliferation with formation of large sinusoidal channels (angioneogenesis) is a typical finding in glioblastomas, and intratumoral hemorrhage is common. The central portions of these tumors frequently outgrow their blood supply and undergo ischemic coagulative necrosis, leaving only a peripheral rim of viable tumor of variable thickness. Cysts of varying size representing the residua of central necrosis may be found within the more solid periphery of many glioblastomas.
The gross pathologic findings described are accurately portrayed on CT and MRI. Although smaller tumors are generally rounded, more extensive lesions have irregular configurations indicating their spread along white matter pathways and penetration into and through cerebral cortex. These tumors typically appear on noncontrast CT as heterogeneous masses with irregular borders of normal or slightly increased density with central cavitation of diminished attenuation (see Fig. 9-14 ). The irregular hyperdense borders demonstrate avid contrast enhancement.
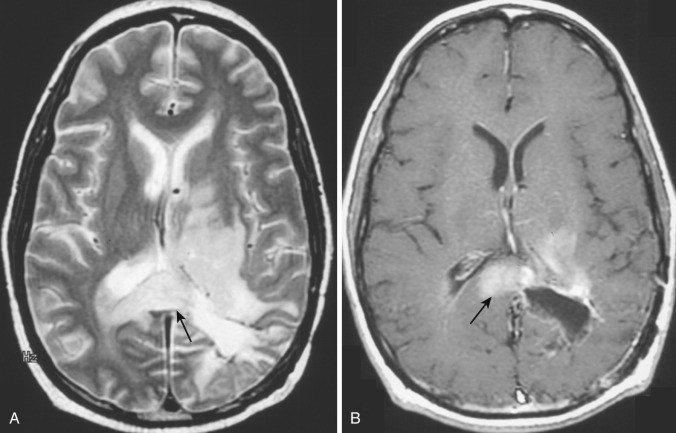
Findings on MRI parallel those on CT. Because of the greater inherent sensitivity of MRI for detecting subacute and chronic hemorrhage and for demonstrating tumor hypervascularity (curvilinear and racemose signal voids), the overall appearance of the lesion is even more heterogeneous on MRI than on CT. Areas of intratumoral central necrosis, which tend to dominate the appearance of the most aggressive of these tumors, appear notably hyperintense on T2-weighted images except where interrupted by more solid masses of cellular debris ( Fig. 9-12A ). Compared with this central necrotic T2 hyperintensity, the surrounding rim of viable and growing tumor appears significantly less hyperintense (owing to its hypercellularity) with unsharp margins and may blend with or be indistinguishable from the peritumoral infiltrative edema.
Virtually all glioblastomas are associated with edema in the surrounding peritumoral white matter, usually graded as moderate or severe ( Figs. 9-13A ; see Fig. 9-15A ). The origin of this edema is uncertain; it may reflect production of a vascular permeability factor by the tumor cells. On CT and on T1-weighted MRI, grossly evident mildly hyperdense or slightly hyperintense tumor margins are typically unsharp, but after IV administration of a contrast agent, they stand out in contrast to the surrounding hypodense or hypointense edematous white matter (see Figs. 9-13B and 9-15B ).
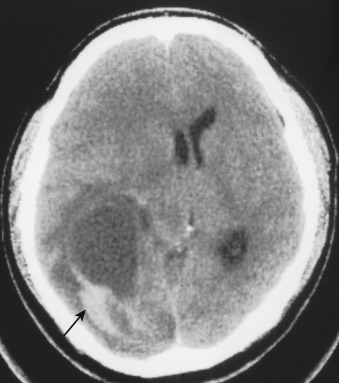
Hemorrhage within the tumor is common in these aggressive tumors with extensive angioneogenesis. The appearance of acute intratumoral hemorrhage in glioblastoma on both CT and MRI differs from acute nonneoplastic intracerebral hemorrhage in that the former is more sharply circumscribed and more heterogeneous in density/signal because of the surrounding and intermixed tumor and necrotic debris ( Fig. 9-14 ), whereas the latter is more irregularly marginated and homogeneous in density/signal because it dissects along otherwise normal white matter tracts. Also, evolution of intratumoral hemorrhage usually proceeds more slowly compared with hypertensive, posttraumatic, or other nonneoplastic types of parenchymal hematomas.
Contrast enhancement of viable tumor, including the peripheral rim and the intratumoral solid portions, is nearly universal in glioblastomas (see Figs. 9-12B, 9-13B, and 9-15B ). The opacifying tumor ring is typically irregular and of varying thickness. The intensity of the enhancement correlates with the level of cellular anaplasia, but it does not accurately reflect the extent of microscopic tumor infiltration. Careful imaging-histopathologic correlation studies in patients with malignant gliomas have conclusively established the presence of viable tumor cells well beyond the margin of contrast enhancement, and the zone of edema in the surrounding white matter must be understood as including foci of microscopic tumor infiltration ; thus the term infiltrative edema.
Differentiation by CT or MRI between glioblastoma (or other grade IV glioma), carcinomatous metastasis, and lymphoma is often difficult. All three processes may appear as irregularly rounded, contrast-enhancing, ringlike masses with walls of varying thickness. In all, the viable tumor rim is typically irregular and nonuniform in thickness, and histologic examination is usually required for definitive diagnosis. Primary CNS lymphoma is typically more central in location and homogeneously solid (see Figs. 9-46 and 9-74 ), but central necrosis, infiltration into the corpus callosum and other major white matter pathways, and tumor extension into the meninges and along the ependyma (all common late manifestations in glioblastoma) are not rare occurrences with lymphoma.
Additionally, a large area of active demyelination with inflammation and edema secondary to previously undiagnosed multiple sclerosis may also simulate a glioblastoma on CT or MRI. Such tumefactive demyelinating lesions (e.g., in multiple sclerosis or in acute disseminated encephalomyelitis [ADEM]) often demonstrate arclike (open toward the adjacent cortex) contrast enhancement and increased or facilitated diffusion, findings that are much less common in glioblastomas.
When images demonstrate multiple nodular or ringlike masses, multicentric glioblastoma must be included in the differential diagnosis ( Fig. 9-15 ), along with metastases and pyogenic cerebral abscesses. The peripheral rim of an abscess is typically (but not always) thinner and more uniform than that of a glioblastoma or a metastasis. Diffusion-weighted imaging (DWI) demonstrates restriction of diffusion in the central cavity of an abscess but not in the coagulative necrosis of a primary or metastatic neoplasm. Besides DWI, other advanced neuroimaging techniques can be used to differentiate glioblastoma, solitary metastases, and abscesses, namely magnetic resonance spectroscopy (MRS), perfusion MRI, and positron emission tomography (PET) imaging, as discussed later in this chapter (see “Advanced Neuroimaging Techniques”).
A commonly encountered clinical problem is the need to differentiate radiation necrosis from recurrent glioblastoma in a patient who has undergone surgical resection and a complete course of radiation therapy and several months after the radiation therapy demonstrates a change in neurologic status. If the follow-up imaging study demonstrates interval increase in the size of the apparent tumor mass and the extent of contrast enhancement, differentiation of recurrent tumor from radiation necrosis in this lesion on the basis of CT or MRI is not possible. In such circumstances, advanced neuroimaging techniques are often employed with perfusion MRI, MRS, and PET imaging, using both 18 F FDG and non-FDG radiotracers, greatly improving sensitivity and specificity, as discussed later in this chapter (see “Advanced Neuroimaging Techniques”).
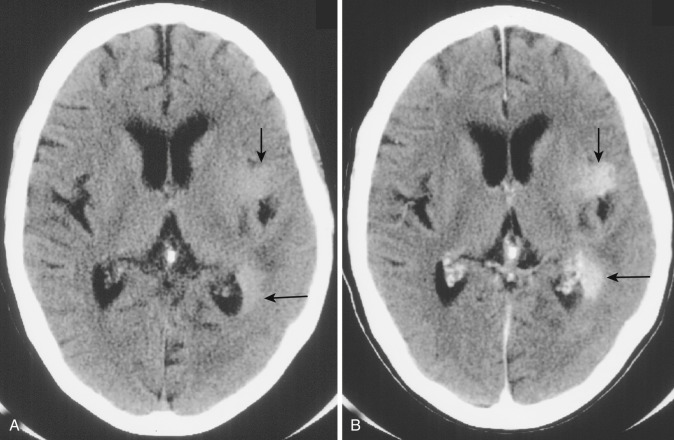
Gliomatosis cerebri.
A comparatively uncommon pattern of glial neoplasia, gliomatosis cerebri is characterized by widespread diffuse neoplastic infiltration in multiple lobes and both cerebral hemispheres without formation of an obvious tumor mass. The cell of origin is uncertain; the tumor is classified as WHO grade III. The extent of infiltration is out of proportion to the other histopathologic features, such as the level of anaplasia and the relative lack of destruction of normal tissues. Despite the extent of tumor infiltration, neural connections are preserved, and the clinical signs and symptoms are relatively mild. Nevertheless the clinical course is relentlessly progressive over periods varying from months to a few years.
The process tends to extend along the white matter tracts but also involves the adjacent gray matter with loss of clear gray-white distinction both on gross pathology and on imaging studies. The involved parenchyma is both expanded and distorted by the tumor infiltration. Microscopically the tumor cells are elongated and infiltrate between myelinated fibers without destroying them, much like in a low-grade astrocytoma, but there is greater cellular atypia and mitotic frequency, and foci of necrosis are occasionally seen.
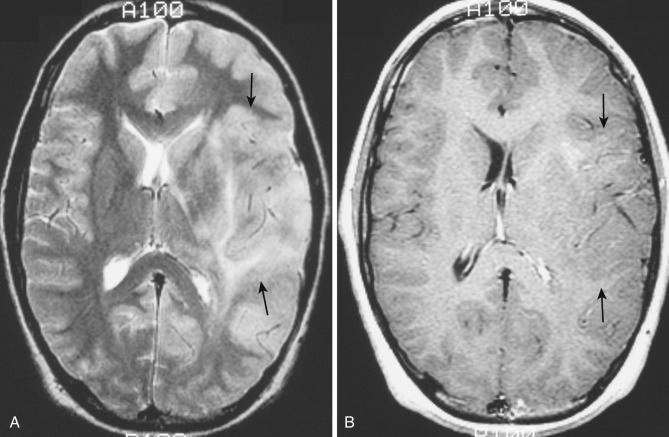
Diagnostic imaging studies demonstrate extensive and scattered areas of parenchymal involvement with ill-defined areas of hyperintensity on T2-weighted MRI and subtle areas of hypodensity on CT scans ( Fig. 9-16A ). No well-demarcated or circumscribed tumor masses are identified on imaging in this disorder. Focal expansion and distortion are seen as areas of diffuse effacement of sulci and compression and displacement of the adjacent ventricles and cisterns. The incidence and level of contrast enhancement are often less than the extent of apparent tumor infiltration (see Fig. 9-16B ) because enhancement relates closely to the degree of cellular dedifferentiation. Clinically the principal differential diagnosis is with multiple sclerosis, but the imaging pattern of multiple widespread areas of expansion and distortion as well as the relatively rapid and consistent progressive clinical course favor a diagnosis of neoplasm.
Gliosarcoma.
A rare tumor, gliosarcoma consists of both glial and mesenchymal elements. Most reported patients have been between 50 and 70 years old. The origin of the mesenchymal element remains uncertain and controversial; it presumably arises from spontaneous neoplastic transformation of vascular elements within the glial neoplasm, but prior radiation therapy has been implicated in a few case reports. The mesenchymal element is usually a fibrosarcoma, but other sarcomas have been reported. Immunohistochemistry studies demonstrate both GFAP reactivity, confirming the astrocytic nature of the glial component, and positive staining for collagen and reticulin, confirming the mesenchymal component.
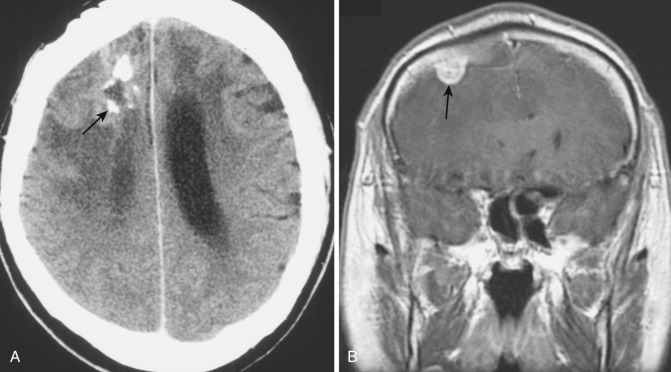
The glial component of these mixed tumors is typically glioblastoma, and gross pathology as well as both CT and MRI demonstrate the variegated pattern of that tumor with areas of central necrosis and peripheral infiltration both along white matter tracts and into adjacent gray matter ( Fig. 9-17 ). Invasion into and involvement of the overlying meninges are often noted (see Fig. 9-17B ); invasion into bone and extracranial soft tissue likely reflects the sarcomatous element within the tumor, and systemic metastasis (a rare occurrence in glioblastoma) is well described for gliosarcoma.
Oligodendroglioma.
Oligodendrogliomas (tumors derived from oligodendrocytes) represent 15% to 20% of intracranial gliomas and 5% to 10% of all intracranial neoplasms. The diagnosis is made much more commonly in recent years than in the past, reflecting a reevaluation of the histologic and immunochemical criteria (oligodendrogliomas are GFAP negative) that define the distinction between oligodendroglioma and astrocytoma. These tumors occur principally in the supratentorial white matter. Like low-grade astrocytomas, they tend to infiltrate along white matter tracts in a nondestructive manner. However, more commonly than astrocytomas, oligodendrogliomas also involve the subcortical white matter and the overlying cortex, and involvement of deep structures is also common. Eighty-five percent occur in the cerebral hemispheres, more than half in the frontal lobes.
The peak age of incidence of oligodendrogliomas is in the fourth and fifth decades of life; these tumors are much less common in childhood or adolescence. The initial clinical presentation is most often a seizure, reflecting the predilection for cortical involvement. Tumor growth is generally slower than in astrocytomas of similar grade (WHO grade II) and the prognosis is better, but a more biologically aggressive category is now recognized in the revised WHO classification as anaplastic oligodendroglioma (WHO grade III). Cytogenetic studies have noted loss of the 1p and 19q chromosomes in approximately half of patients with oligodendroglioma; on imaging studies, patients with loss of 1p and 19q have demonstrated a significant association with positive response to multiagent nitrosourea-based chemotherapy.
Oligodendrogliomas are typically densely cellular and solid, contain multiple irregular masses of dystrophic calcification, and grow very slowly. Median survival is approximately 10 years. They can attain considerable size before they produce symptoms. They may contain a dense network of branching capillaries. Areas of cystic degeneration are found in 20% of cases, mainly in the larger tumors. Intratumoral hemorrhage is also a frequent finding. Treatment is primarily surgical resection with adjuvant chemotherapy and radiotherapy, but local recurrence is common.
Anaplastic oligodendrogliomas on histology demonstrate greater cellular atypia and mitotic activity, more extensive tumor vascularity, and necrosis. The prognosis is less favorable, with median survival of 4 years and 10-year survival of approximately 15%.
Foci of astrocytoma are frequently found in association with oligodendroglioma, and when these are prominent, the tumor is termed an oligoastrocytoma. Oligoastrocytomas are typically WHO grade II tumors and anaplastic oligoastrocytomas WHO grade III. When there is a clear preponderance of neoplastic oligodendrocytes (75%-90%), the tumor is classified as an oligodendroglioma.
On CT, oligodendrogliomas most often appear as masses of heterogeneous density (predominantly hypodense) in the frontal lobe, with irregular and poorly defined margins involving the more superficial white matter and frequently infiltrating into the overlying cortex and obscuring the gray-white interface. The characteristic feature in the great majority of cases is the presence of large nodular or clumplike calcifications within the tumor ( Fig. 9-18A ). The frequency of intratumoral calcification in oligodendrogliomas (70%-90%) is the highest of any cerebral neoplasm, but overall, intratumoral calcification is most common in astrocytomas (frequency, 25%) because of the much greater prevalence of that lesion. Intratumoral cyst formation is common in oligodendrogliomas (20%), but frank intratumoral necrosis is unusual and indicates a poor prognosis. Contrast enhancement is noted in approximately 50% of oligodendrogliomas (see Fig. 9-18C ), but the degree of enhancement is variable and appears to correlate with higher histologic grade and lower survival.
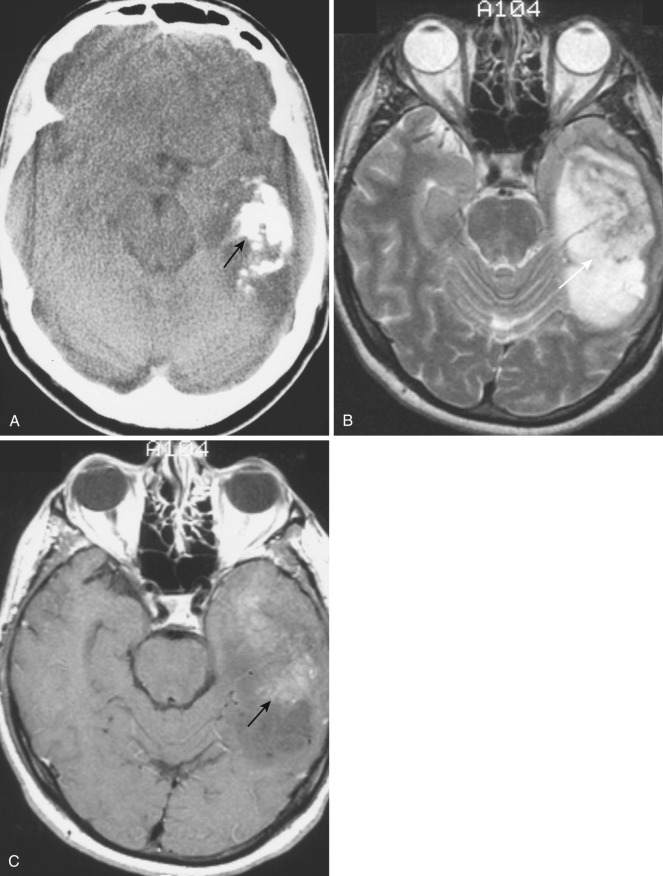
On MRI, oligodendrogliomas appear heterogeneous on both T1- and T2-weighted images because of the presence of intratumoral cysts and calcifications. On T1-weighted images, the tumors appear predominantly hypointense to gray matter, and on T2-weighted images they are most often hyperintense, with small intratumoral cysts and focal calcifications and hemorrhages contributing to the overall heterogeneity (see Fig. 9-18B ). Peritumoral edema is noted in about one third of cases. Enhancement is variable and typically heterogeneous, with anaplastic oligodendrogliomas (grade III) enhancing more commonly than low-grade oligodendroglioma (grade II). Also, as with other gliomas, new enhancement within a previously nonenhancing oligodendroglioma suggests anaplastic transformation.
Ependymoma.
The ependyma lining the floor of the fourth ventricle is the most common site of origin of intracranial ependymomas (65%). Nevertheless, approximately 30% of ependymomas occur above the tentorium, arising mainly from ependymal cell rests in the cerebral white matter adjacent to the lateral ventricles. Infratentorial ependymomas occur predominantly in children between 1 and 6 years of age; supratentorial ependymomas occur mainly in the second through the fourth decades of life.
Posterior fossa ependymomas make up approximately 10% to 15% of all primary CNS neoplasms of childhood and approximately 3% to 5% of all intracranial neoplasms. They are the third or fourth most common posterior fossa tumor of childhood, exceeded only by PA, medulloblastoma, and possibly brainstem glioma. Ependymomas are usually slow-growing tumors of moderate malignancy (WHO grade II), but the prognosis is guarded because of their notable tendency to recur locally and disseminate via the subarachnoid spaces. The incidence of seed metastases via the CSF is approximately 10% overall but is considerably higher in the infratentorial tumors of early childhood; tumor seeding into the subarachnoid spaces is found initially or on follow-up studies in approximately one third of fourth ventricle ependymomas.
Supratentorial ependymomas are commonly paraventricular, located most frequently within cerebral parenchyma near the atrium of the lateral ventricle or the foramen of Monro and presumably arising from ependymal cell rests. Infratentorial ependymomas are lobulated, well-circumscribed, soft exophytic masses that expand within the cavity of the fourth ventricle rather than infiltrate into the surrounding parenchyma. They often extend in a tonguelike fashion through the lateral recesses into the lateral medullary and cerebellopontine angle cisterns (15%) or through the foramen of Magendie into the vallecula (10%). Tumor growth within the fourth ventricle eventually leads to occlusion of the ventricle, with resultant obstructive hydrocephalus.
Ependymomas are highly cellular with small uniform cells that tend to form characteristic perivascular pseudorosettes. Although they do not invade through the ventricular walls, they are firmly attached to the ventricular floor, and complete resection of the tumor base is frequently not possible. The rate of local tumor recurrence after surgical resection is 90% to 95% within 3 years, and the 5-year survival is currently 45% to 70%. Furthermore, about 25% of ependymomas exhibit features of anaplasia, with a high mitotic rate, cellular pleomorphism, and intratumoral necrosis; these tumors are known as anaplastic ependymomas (WHO grade III).
Intratumoral calcification and cyst formation are common in ependymomas (20%-50% of reported cases). Peritumoral edema is a prominent and frequent finding in association with both cerebellar and cerebral ependymomas.
On noncontrast CT, fourth ventricle ependymomas appear as hypodense or isodense heterogeneous, well-circumscribed, rounded masses within the fourth ventricle that partially or completely obliterate the cavity of the ventricle. The tumor is typically well defined by a prominent surrounding hypodense halo of peritumoral edema. Aggregates of calcification, usually small and round but sometimes quite large, are found in up to half of these lesions, and focal lucencies resulting from cyst formation are nearly as common. In supratentorial tumors, the cysts are both larger and more frequent (up to 80%) than in fourth ventricle ependymomas (see Fig. 9-20A ). Intratumoral hemorrhage is relatively less common, with reported frequency of 20% or less. After IV administration of contrast agent, the solid portions and cyst walls of the tumor mass exhibit moderate but variable heterogeneous contrast enhancement.
The MRI appearance of ependymomas is heterogeneous, reflecting mixed signals from intratumoral calcification, cysts, and hemorrhage. Usually these tumors are isointense to hypointense relative to white matter on T1-weighted images and isointense to hyperintense to white matter on T2-weighted images ( Fig. 9-19A and B ; Fig. 9-20B ). Contrast enhancement is the rule, but it is variable in degree and typically not uniform, seen in both ependymoma and anaplastic ependymoma. (see Figs. 9-19C and 9-20C ). Although the MRI features of ependymoma are nonspecific, the multiplanar capabilities of this modality offer unique visualization of infratentorial tumor extent within the cranial vault and the region of the cervicocranial junction.
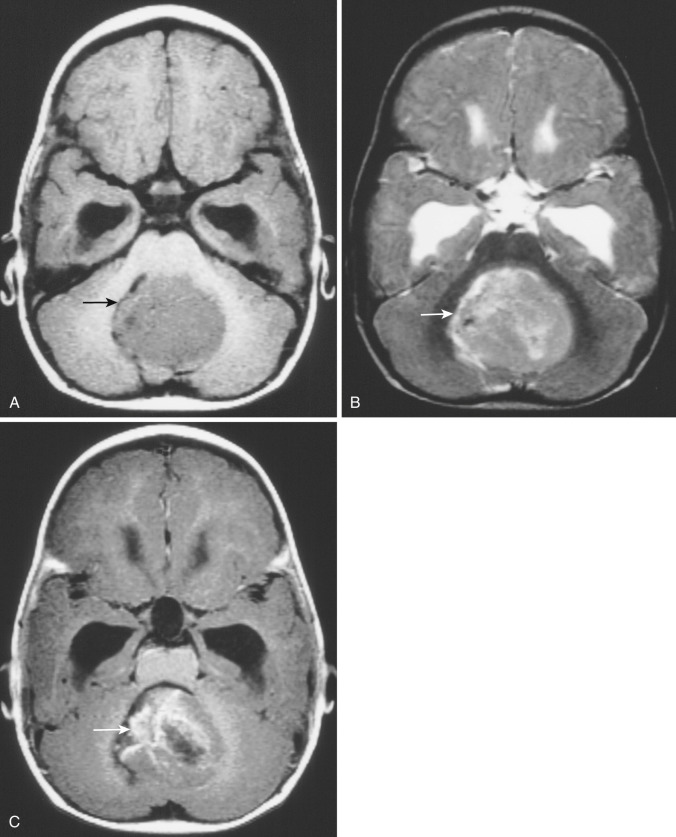
On both CT and MRI as well as in the clinical setting, the presentation of ependymoma of the fourth ventricle may closely resemble that of medulloblastoma and PA. The incidence and extent of intratumoral calcification, heterogeneity of density or signal intensity, and propensity for tumor extension into the fourth ventricular recesses are greater in ependymomas than in medulloblastomas or astrocytomas. In general, medulloblastomas appear more dense on CT with a lower frequency of intratumoral calcification, and they exhibit more homogeneous contrast enhancement than ependymomas. Differentiation can be difficult but is aided by DWI, as discussed later in this chapter. Astrocytomas tend to occur in slightly older patients and are much less frequently centered in the midline.
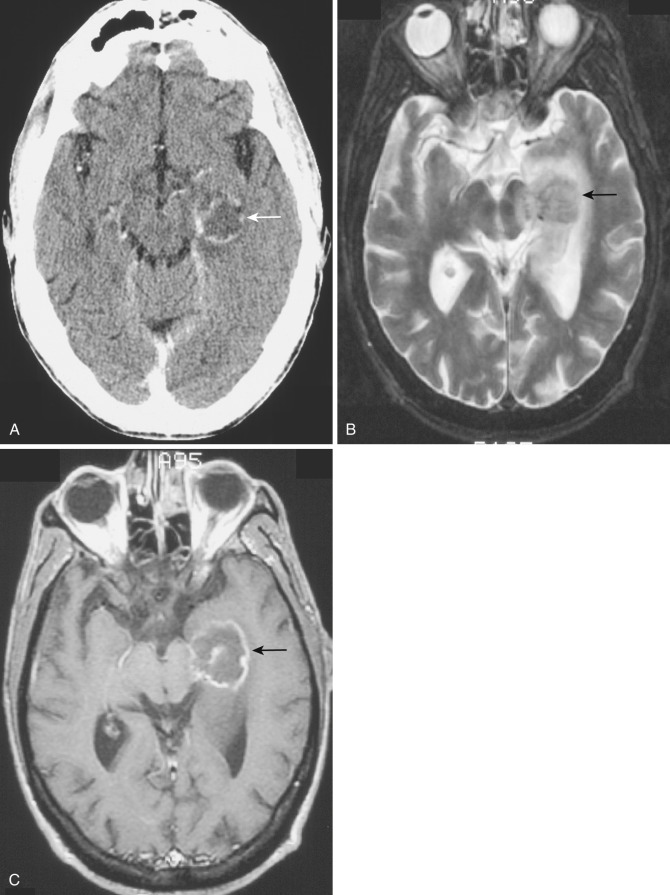
Subependymoma.
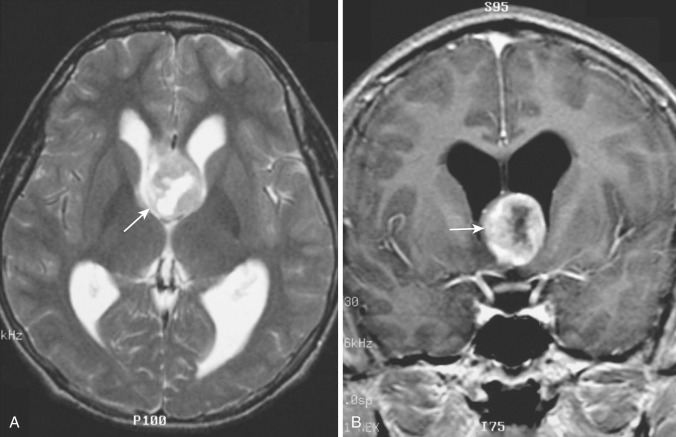
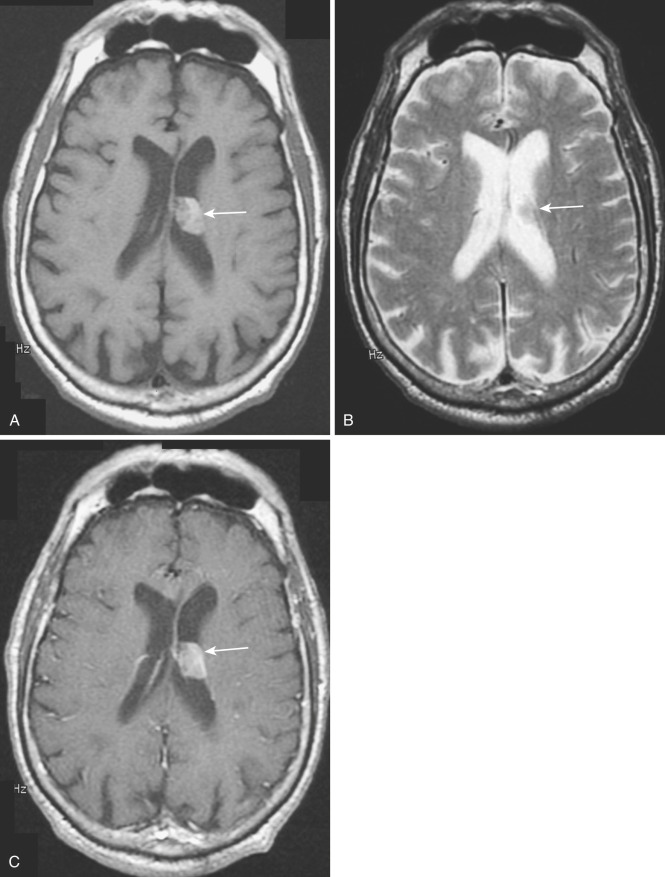
A rare variant of ependymoma that actually consists of both highly differentiated ependymal cells and astrocytes, subependymoma is classified as a benign (WHO grade I) glial neoplasm. It is considered to arise from beneath the ventricular lining and projects into the ventricular cavity as a sharply outlined intraventricular mass but does not extend deeply from its base into the adjacent brain parenchyma ( Fig. 9-21 ). Subependymomas are typically solid and homogeneous, although larger tumors may contain calcifications, microcystic changes, and evidence of intratumoral hemorrhage.
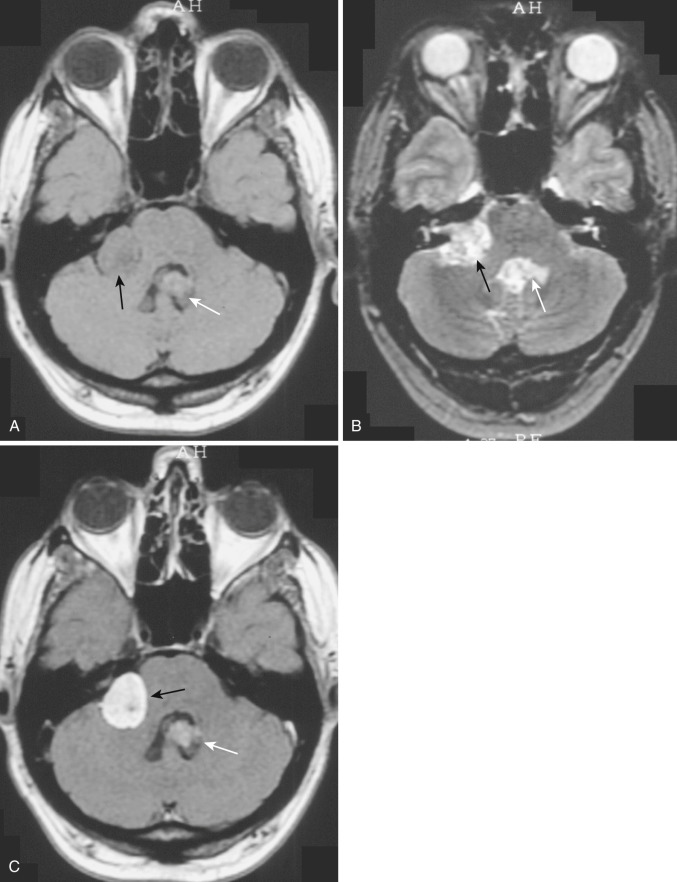
Subependymomas occur most commonly (50%-60%) in the fourth ventricle but also arise in the lateral ventricles. Most present clinically in adults, but many of these lesions are asymptomatic and found incidentally at autopsy. Those that become symptomatic (causing obstructive hydrocephalus) occur mostly in the lateral ventricles ( Fig. 9-22 ) in relation to the septum pellucidum and foramen of Monro and much less commonly in the region of the aqueduct.
On CT and MRI, symptomatic lesions in the fourth or lateral ventricles usually appear homogeneous in density and signal intensity. They appear homogeneously isodense or slightly hypodense on CT. On T2-weighted MRI the tumor appears homogeneously hyperintense to adjacent brain. There is variable contrast enhancement after IV administration of contrast medium. However, larger tumors may exhibit internal heterogeneity of density and signal owing to intratumoral calcification and hemorrhage.
The differential diagnosis includes central neurocytoma, ependymoma, and subependymal giant cell astrocytoma. Patient age, nature of the symptoms, presence of associated disease, tumor size, presence and extent of calcification, cystic change, evidence of hemorrhage, and contrast enhancement are the key factors in making the differentiation.
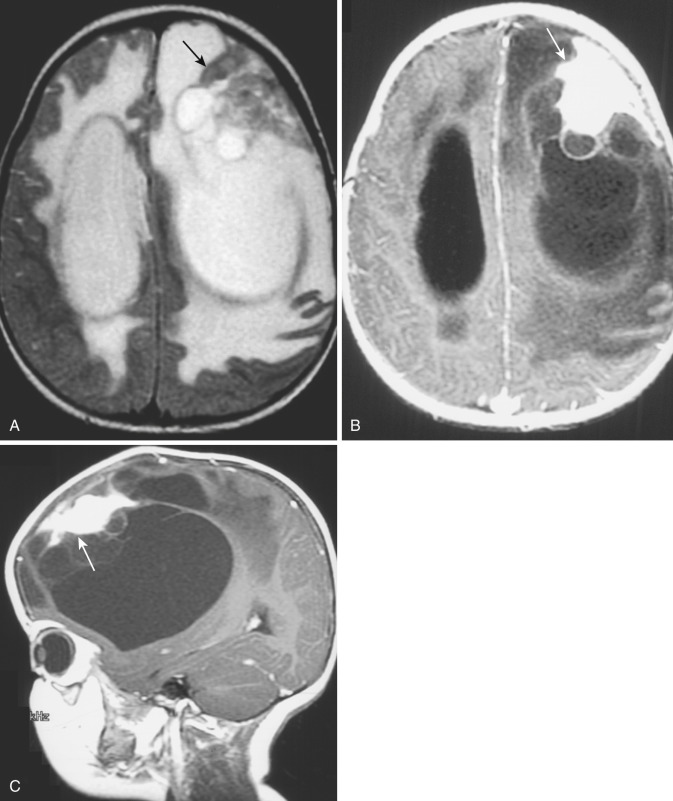
Choroid Plexus Papilloma and Carcinoma.
Papillomas arising in the choroid plexus are benign intraventricular neoplasms that locally expand the involved ventricle and cause hydrocephalus, which may be secondary to ventricular obstruction, overproduction of CSF, or impairment of CSF absorption. Choroid plexus papillomas represent less than 1% of all brain tumors but are more common in young children (3% of all pediatric brain tumors), particularly in the neonatal period; 85% occur in the first 5 years of life, and these tumors represent 40% of all brain tumors in the first 60 days of life.
The atria of the lateral ventricles and fourth ventricle are the most common sites of involvement. An age disparity exists with regard to tumor location, with lateral ventricle tumors occurring mainly in young children, and fourth ventricle papillomas appearing more commonly in adults. It is not uncommon for fourth ventricle tumors to extend through the lateral recesses and protrude into the cerebellopontine angle cisterns.
Histologically, choroid plexus papillomas consist of well-differentiated proliferation of both the surface epithelium of the choroid plexus and the underlying vascular connective tissue core and are usually classified as WHO grade I. They grow slowly and are noninvasive, tending to expand within the confines of the ventricle or its outlet foramina. Occasionally, however, the epithelium may exhibit malignant change, and in such cases the tumors are classified as carcinomas; they may extend into the adjacent brain parenchyma. The prognosis in these malignant choroid plexus tumors depends on the completeness of surgical resection, but overall the 5-year survival ranges from 26% to 40%.
Because of the friability and high vascularity of choroid plexus papillomas, intratumoral and intraventricular hemorrhage are common.
On CT, choroid plexus papillomas appear as large, rounded or lobulated, isodense or hyperdense intraventricular masses that engulf and separate the normal choroid plexus calcifications and exhibit intense homogeneous contrast enhancement. Small hypodense nonenhancing foci are occasionally identified within these tumors.
On MRI, choroid plexus papillomas are usually isointense or hypointense with respect to brain on T1-weighted images and heterogeneously hyperintense relative to brain on T2-weighted images ( Fig. 9-23A and B ) ; the heterogeneity of signal intensity within the tumor mass may be due to areas of vascularity, calcification, or previous hemorrhage (see Fig. 9-23A ). As noted previously the tumor typically expands the ventricle locally at the site of origin and may obstruct ventricular drainage, causing dilatation (entrapment) of the more proximal portion of the affected ventricle. On both CT and MRI, these tumors usually demonstrate intense contrast enhancement (see Fig. 9-23C ), which may be homogeneous or heterogeneous depending on the tumor contents.
The differential diagnosis of choroid plexus papilloma on imaging studies usually includes ependymoma in older children and adults and meningioma in adults. The lobulated tumor margins, lack of invasion of brain parenchyma despite the large tumor size, and heterogeneous signal pattern within the tumor all aid in distinguishing this lesion.
Nonglial Tumors
Neuronal and Mixed Neuronal and Glial Tumors.
In this group of tumors of neuroepithelial origin, neuronal and glial differentiation is present in varying degrees. This chapter will discuss the following recognized entities: ganglioglioma, gangliocytoma, dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease), desmoplastic infantile ganglioglioma, dysembryoplastic neuroepithelial tumor, and central neurocytoma.
Ganglioglioma and gangliocytoma.
Gangliogliomas are uncommon low-grade tumors consisting of a mixture of neoplastic astrocytes and dysplastic neurons. Slow-growing neoplasms with well-differentiated ganglion cells and a low-grade astrocytomatous stroma, these tumors are considered WHO grade I or II. Gangliocytomas (also known as ganglioneuromas ) are relatively less common and contain only neuronal elements; they are classified as WHO grade I. Together these tumors account for 1.3% of all primary brain tumors.
Gangliogliomas and gangliocytomas occur in young people (peak incidence in the second decade of life) and constitute approximately 3% to 4% of primary brain tumors in children. They are found most commonly in the temporal lobes but may occur anywhere in the cerebral hemispheres. The clinical presentation is usually with seizures of long duration, and gangliogliomas are considered the most common cause of chronic temporal lobe epilepsy. These tumors are usually well-circumscribed solid masses but may include one or more cysts. Gross total resection of a tumor is the treatment of choice. Overall, gangliogliomas and gangliocytomas have a 92% 3-year survival. In approximately 10% of cases, however, the tumor is more aggressive; in these cases, the malignant element is always glial.
Diagnostic imaging findings are relatively nonspecific and similar to those in other low-grade intracerebral neoplasms. Noncontrast CT scans demonstrate relatively well-circumscribed hypodense (occasionally isodense with adjacent brain) lesions typically located in the periphery of the hemisphere, with little associated mass effect or surrounding vasogenic edema. Foci of calcification are identified in 33% to 40% of cases, and cysts in 50% of cases. Calcification is less common in noncystic tumors. Rarely a peripherally located indolent tumor may erode the inner table of the overlying skull. Hemorrhage is rare in these tumors. Contrast enhancement is observed in about 50% of cases and may be homogeneous or heterogeneous depending on the presence and size of cystic and calcific changes.
On MRI, these tumors (like many other intracerebral neoplasms) are usually heterogeneous in appearance, predominantly hypointense to gray matter on T1-weighted images and hyperintense on T2 ( Fig. 9-24A ). Mild to moderate contrast enhancement of the solid noncystic portion of the tumor is found in the majority of cases on both CT and MRI (see Fig. 9-24B ).
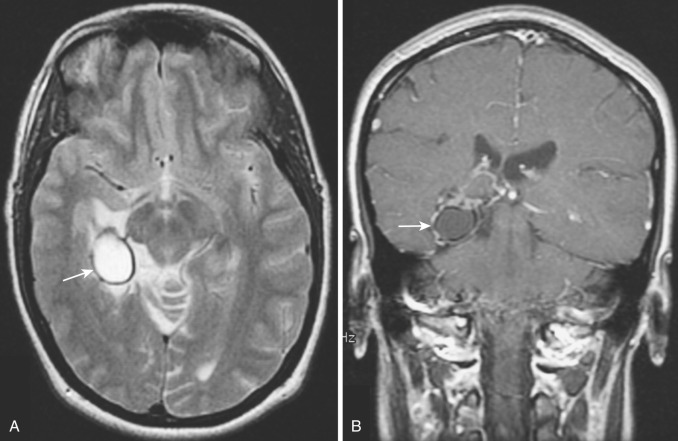
Findings on CT and MRI in these ganglion cell tumors are not characteristic; oligodendroglioma is a common differential consideration. However, in a young patient with a protracted history of seizures and a relatively well-circumscribed cystic tumor containing calcification in the periphery of the temporal lobe with little or no associated mass effect, ganglioglioma should be strongly considered.
Dysplastic gangliocytoma of the cerebellum.
A rare lesion, dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) is considered a complex hamartoma or malformation and is classified as WHO grade I. Dysplastic gangliocytoma may exist as an isolated entity or as part of Cowden’s disease, a rare syndrome with multiple mucosal hamartomas. The clinical presentation is usually ataxia and signs of increased intracranial pressure in a young adult. A large dysplastic mass occupies most or all of the cerebellar hemisphere, with thickening of the granular and molecular layers of the overlying cerebellar cortex ( Fig. 9-25A ). The enlarged mass causes displacement and compression of the fourth ventricle. On MRI the mass is poorly demarcated, hypointense on T1-weighted images and hyperintense on T2-weighted images, and the thickened folia present a distorted striate pattern (see Fig. 9-25B ). Contrast enhancement of either the tumor or the thickened cerebellar folia is unusual (see Fig. 9-25C ).
Desmoplastic infantile ganglioglioma.
Desmoplastic infantile ganglioglioma (DIG) is an unusual lesion that occurs in young infants. It consists of immature neurons and astrocytes together with extensive fibrocollagenous (desmoplastic) thickening and cyst formation. The solid desmoplastic portion is often located adjacent to the meninges. Despite its variegated appearance, this tumor is classified as WHO grade I and has a favorable long-term prognosis after complete excision.
DIGs are large and superficial hemispheric tumors that occur mainly in the frontal and parietal lobes and present a markedly heterogeneous appearance on both gross inspection and MRI ( Fig. 9-26 ). The solid portions of this tumor are hypointense relative to normal brain on T2-weighted images and frequently demonstrate intense contrast enhancement. DIG differs from the usual ganglioglioma in that it presents in infancy, is predominantly frontoparietal in location, and has dense desmoplasia. On MRI, the intermixed collagenous and multicystic pattern of this lesion most resembles that of a primitive neuroectodermal tumor (PNET), which carries a much less favorable prognosis; however, PNET is also characterized by the common presence of intratumoral calcification, hemorrhage, and necrosis, findings that are uncommon in DIG.
Dysembryoplastic neuroepithelial tumor.
Dysembryoplastic neuroepithelial tumors (DNETs) are uncommon benign intracortical tumors that characteristically occur in young patients (<20 years) above the tentorium, mainly in the temporal lobe (60%-80%) or frontal lobe. They can be multifocal and rarely arise in the septum pellucidum. Patients typically present with intractable partial seizures. Partial or complete excision is the treatment of choice, and no recurrences have been reported. The tumor is termed dysembryoplastic because of its multinodularity, with areas of cortical dysplasia at the margins between the tumor nodules and adjacent brain.
DNETs are lesions of long standing that most frequently involve the convexity cortex and often protrude beyond the adjacent cortical margin, eroding the overlying inner table of the calvarium. The imaging findings, except for the superficial cortical location, are similar to those of other low-grade glial tumors (astrocytoma, oligodendroglioma). On CT, DNET typically appears as a well-circumscribed hypodense superficial mass, occasionally with intratumoral calcification or cystic change or both. MRI demonstrates a mass centered in the convexity cortex and bulging externally that is hypointense to adjacent brain on T1-weighted images ( Fig. 9-27B ) and hyperintense on T2-weighted images (see Fig. 9-27A ) with no surrounding edema. The protruding external margin may present a “soap bubble” appearance, reflecting internal cystic change. Contrast enhancement is seen in only a minority of these lesions (see Fig. 9-27C ).
Central neurocytoma.
A relatively rare benign slow-growing (WHO grade II) intraventricular tumor, central neurocytoma has now been recognized as a separate entity. It occurs almost exclusively in the anterior portion of the lateral ventricle, arising from the superolateral ventricular wall and extending medially adjacent to the septum pellucidum and the foramen of Monro; extension through the foramen into the third ventricle is unusual. No such tumors have been reported arising in the third or fourth ventricles, and only a few hemispheric lesions have been found. Central neurocytomas are tumors of young adults and are rare in children and older adults. Patients typically present clinically with headache and signs of increased intracranial pressure secondary to ventricular obstruction. Partial or complete surgical excision is the treatment of choice, and no deaths from tumor growth or recurrence have been reported.
Histologically, central neurocytoma is identical to oligodendroglioma. It is characterized by nests of small well-differentiated cells separated by thin fibrovascular septa. Intratumoral calcification and multiple small cysts are common findings, but hemorrhage (either intratumoral or intraventricular) is unusual. It is only with electron microscopy and immunohistochemistry that its neuronal origin can be determined.
CT demonstrates a sharply marginated, inhomogeneous, isodense to slightly hyperdense intraventricular mass with a broad-based attachment to the wall of the frontal horn or anterior body of the lateral ventricle and abutting the septum pellucidum. Calcifications, either coarse or globular, and multiple small intratumoral hypodense cysts are found in 50% or more of central neurocytomas. Mild to moderate contrast enhancement of the isodense to hyperdense regions is common.
The findings on MRI are similar. The tumor demonstrates a heterogeneous pattern with intermixed foci of isointensity (compared with adjacent gray matter) and hypointensity (due to calcifications and cysts) on T1-weighted images and isointensity to hyperintensity on T2-weighted images ( Fig. 9-28A ). Inhomogeneous mild contrast enhancement is common (see Fig. 9-28B ).
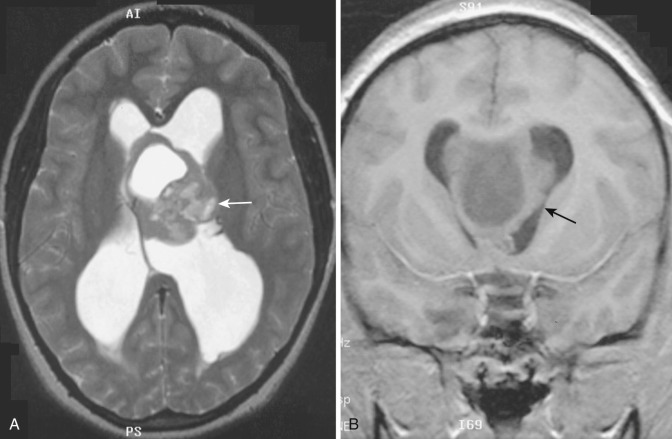
The differential diagnosis is that of a well-circumscribed intraventricular mass at or near the foramen of Monro and includes subependymal giant cell astrocytoma and subependymoma. The absence of deep extension into adjacent brain parenchyma, associated nodular tubers and intratumoral hemorrhage, and the relatively young age of the patient favor the diagnosis of central neurocytoma.
Pineal Parenchymal Tumors.
Tumors arising within the parenchyma of the pineal gland constitute less than 1% of all primary brain tumors in adults. However, pineal tumors represent nearly 10% of all pediatric brain tumors. The most common tumors to involve the pineal gland are the germ cell tumors, which constitute 50% to 70% of pineal region tumors and are discussed elsewhere in this chapter.
Pineal parenchymal tumors represent only 15% to 30% of pineal region neoplasms. They include a range from primitive undifferentiated (pineoblastoma) through transitional forms (primary parenchymal tumor of intermediate differentiation) to well-differentiated (pineocytoma).
Pineoblastoma.
Pineoblastomas are pediatric tumors with a peak incidence in the first 2 decades of life and rare after age 30. These malignant highly cellular undifferentiated small cell tumors are histologically and histochemically identical to other primitive neuroectodermal tumors and exhibit similar biological behavior. They are locally invasive and may spread into the third ventricle, thalami, and quadrigeminal plate. Pineoblastomas frequently contain areas of hemorrhage and necrosis, and intratumoral calcification is common. Presenting symptoms include precocious puberty (possibly related to destruction of normal pineal tissue with loss of melatonin secretion), Parinaud’s syndrome and other cranial nerve deficits, and obstructive hydrocephalus. Dissemination via the CSF with leptomeningeal and ependymal metastases occurs in 10% of cases and may be found at the time of initial diagnosis. The 5-year survival rate is 58%.
On CT, pineoblastomas appear hyperdense with irregular intratumoral calcification, infiltration into neighboring structures, and mass effect on the posterior aspect of the third ventricle and the tectum of the midbrain. They demonstrate dense contrast enhancement. On MRI, pineoblastomas appear heterogeneous in signal, predominantly hypointense to gray matter on T1-weighted images and isointense to gray matter on T2-weighted images, with interspersed foci of hypointensity secondary to intratumoral necrosis and calcification ( Fig. 9-29A and B ). As on CT, contrast enhancement of the solid portion of the tumor is the rule (see Fig. 9-29C ). The chief differential consideration is germinoma, which may look identical on CT and MRI.
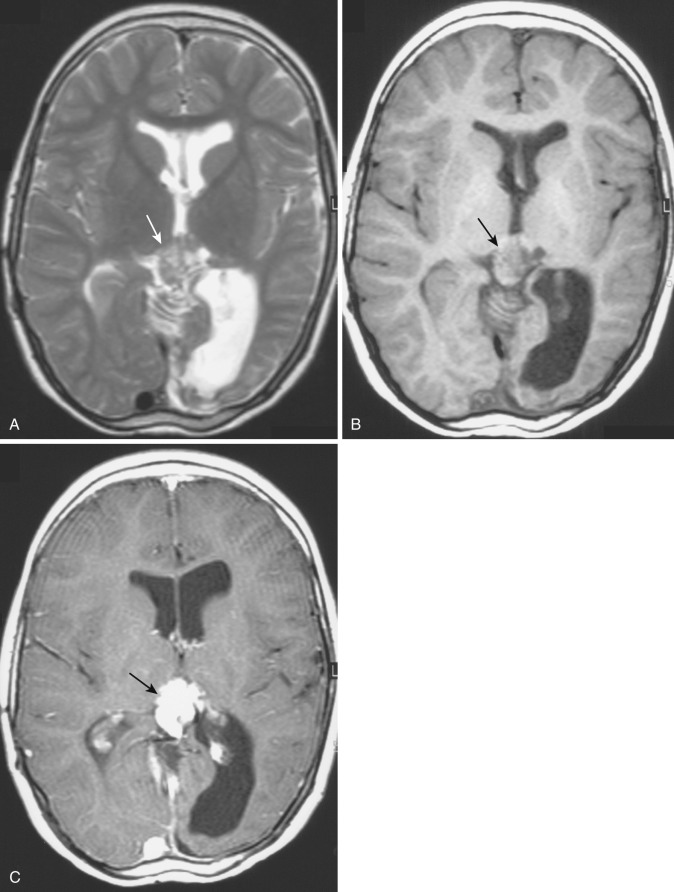
A rare occurrence that is nevertheless important for recognition and management is the association of bilateral retinoblastomas with pineoblastoma, often termed trilateral retinoblastoma. This condition, an inherited disorder occurring in very young children (<2 years), is found in 3% of patients with bilateral retinoblastomas.
Pineocytoma.
Pineocytomas are tumors of adults, with a mean age of presentation of 36 years. Compared with pineoblastomas, these lesions are less densely cellular with well-differentiated cells that have more cytoplasm arranged in lobules in an architecture similar to that of the normal pineal gland. They tend to follow a slower and more benign clinical course, but transitional tumors intermediate in cellularity and mitotic activity also exist. Intratumoral calcification is present in more than 50%. Subarachnoid seeding is not seen in the more benign neoplasms. The 5-year survival for pineocytomas is 67%.
On CT, the typical appearance is of a rounded, well-circumscribed, homogeneously hyperdense lobulated expansile mass with intratumoral calcification situated at and often obstructing the posterior margin of the third ventricle ( Fig. 9-30A ). There is no evidence of invasion of neighboring structures. Multiplanar MRI demonstrates a rounded or ovoid mass compressing the posterior margin of the third ventricle and the superior aspect of the quadrigeminal plate. The mass is homogeneously hypointense to isointense on T1-weighted images and hyperintense relative to gray matter on T2-weighted images. The signal characteristics reflect the higher water content of these more mature tumors compared with pineoblastomas. Contrast enhancement of all or a major portion of a pineocytoma is the rule on both CT (see Fig. 9-30B ) and MRI.
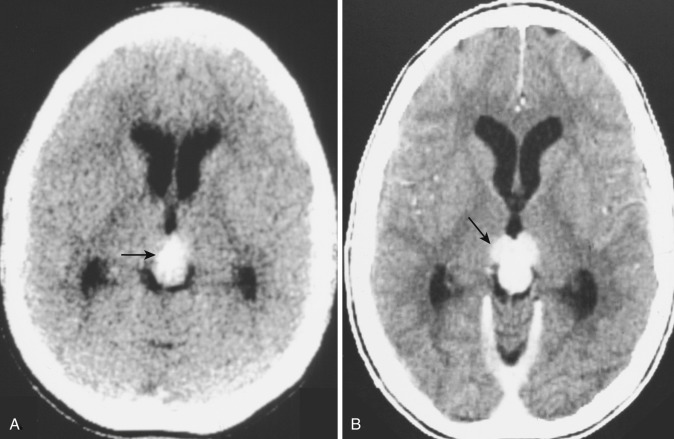
Embryonal Tumors.
The terminology regarding primitive or embryonal tumors of neuroepithelial origin has been a source of controversy for more than 2 decades. Gradually, however, a consensus appears to be emerging concomitant with advances in applied molecular biology. PNETs of the CNS appear to be one group of a larger category of embryonal tumors that occur throughout the body (e.g., neuroblastomas of the adrenal and sympathetic ganglia). These are the second most common intracranial tumors of childhood, representing approximately 15% to 20% of all childhood brain tumors and exceeded in frequency only by astrocytomas. They have a common histology consisting of densely packed undifferentiated round cells with a high nucleus-to-cytoplasm ratio and high mitotic activity but follow divergent patterns of differentiation.
The classic PNET is the medulloblastoma of the cerebellar vermis, which constitutes 85% of this group of tumors. The remaining 15%, almost all supratentorial, include pineoblastomas (see earlier), ependymoblastomas, and medulloepitheliomas. The histogenesis of PNETs is undetermined, but one hypothesis is that they arise from immature pluripotential precursor cells in the subependymal layers of the developing ventricular system and in the external granular layer of the cerebellum.
Medulloblastoma.
An invasive and highly malignant tumor (WHO grade IV), medulloblastoma is the second most common primary brain tumor of childhood and the most common pediatric posterior fossa neoplasm. Although medulloblastomas may occur at any age, 75% present clinically before age 15. There are two distinct age peaks, at 4 to 8 years and 15 to 35 years. In children, medulloblastomas are primarily tumors of the cerebellar vermis ( Fig. 9-31 ), but in adults they tend to be located more laterally in the cerebellar hemispheres (see Fig. 9-31 ). The typical clinical presentation consists of signs of obstructive hydrocephalus, cerebellar dysfunction, or both—headache, nausea, vomiting, and ataxia.
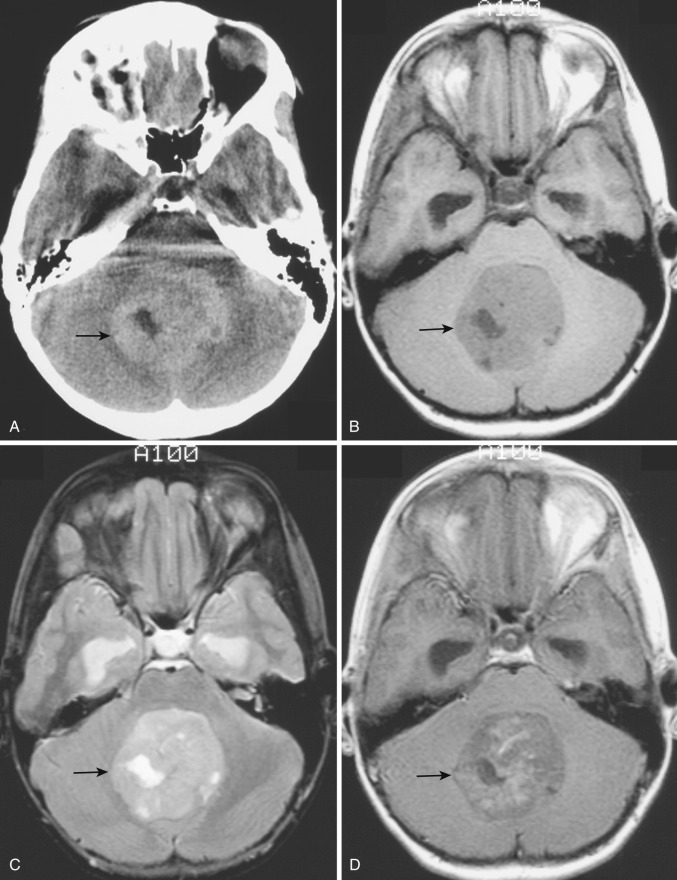
These aggressive tumors grow rapidly with extension from the vermis into the fourth ventricle and spread inferiorly into its recesses and outlet foramina and superiorly into the aqueduct. More than 90% present clinically with hydrocephalus. They have a strong tendency to seed via the CSF, and approximately 40% demonstrate subarachnoid or retrograde ependymal metastases at the time of initial diagnosis. They are highly radiosensitive, and the standard treatment approach is gross total resection plus chemotherapy and total craniospinal axis irradiation. Ten-year survival rates are in the range of 50% to 70%, but the prognosis is poorer in children younger than 3 years. Recurrence after the initial treatment regimen is about equally distributed between local regrowth at the primary tumor site and distant metastasis. Systemic metastasis, a highly unusual occurrence with most CNS primary neoplasms, is identified in approximately 5% of patients, mainly to bone.
Compared with other highly aggressive brain tumors, medulloblastomas appear relatively discrete and solid with convex margins on gross inspection but may contain foci of necrosis and hemorrhage. Microscopically the tumor cells tend to be arranged in pseudorosettes, and areas of vascular proliferation, cellular pleomorphism, and calcification are occasionally identified. Electron microscopy and immunostaining usually confirm neuronal differentiation. Approximately 15% of medulloblastomas contain a significant amount of collagen and reticulin and may demonstrate a nodular pattern; designated desmoplastic medulloblastomas, these tumors are relatively more common in the cerebellar hemispheres in adolescents and young adults.
On noncontrast CT, medulloblastomas appear as rounded or oblong, mainly homogeneous, isodense to slightly hyperdense masses centrally located in the inferior vermis and cavity of the fourth ventricle (see Fig. 9-31A ). Differentiation of medulloblastoma from diffuse solid cerebellar astrocytoma is most reliably made on noncontrast CT. The solid hyperdense appearance of medulloblastoma reflects the dense cellular nature of this tumor, whereas astrocytomas are typically rather uniformly hypodense. Obstruction of the fourth ventricle, inferior aqueduct, or both with resulting enlargement of the third and lateral ventricles is commonly seen on CT at the time of clinical presentation. Punctate or nodular intratumoral calcifications are identified in 10% to 20% of cases. Hypodense intratumoral foci of necrosis and cysts are found in up to 50% of medulloblastomas (see Fig. 9-31A ). Many medulloblastomas are surrounded by a thick poorly marginated hypodense halo representing extensive peritumoral edema (see Fig. 9-31A and B ). The imaging findings in young adults are similar except for the more lateral (hemispheric) location of the tumor.
After IV administration of contrast medium, moderate to intense homogeneous enhancement of the solid tumor mass is the rule in medulloblastomas, although approximately 10% do not demonstrate contrast enhancement on CT. Areas of tumor seeding in the posterior fossa cisterns or in the third or lateral ventricles exhibit similar homogeneous contrast enhancement in a nodular or sheetlike configuration.
The appearance of medulloblastomas is less specific on MRI than on CT. The tumors are mildly hypointense to isointense relative to the adjacent cerebellar vermis on T1 and isointense to slightly hyperintense on T2-weighted images ( Fig. 9-32A ; see Fig. 9-31B and C ). Isointensity (or even hypointensity) of the tumor matrix on T2-weighted images is likely due to the hypercellularity and high nucleus-to-cytoplasm ratio of this tumor. However, heterogeneity of signal intensity is often seen on T2-weighted images (see Figs. 9-31C and 9-32B ) secondary to intratumoral cysts, necrosis, vascular flow voids, and hemorrhage. Contrast enhancement of the solid portions of the tumor, seen in more than 90% of patients, is typically intense and homogeneous (see Figs. 9-32B and 9-75A ) but may be irregular and patchy (see Fig. 9-31D ). MRI with contrast enhancement is very sensitive for detecting tumor spread and metastatic seeding in the cranial and spinal subarachnoid spaces.
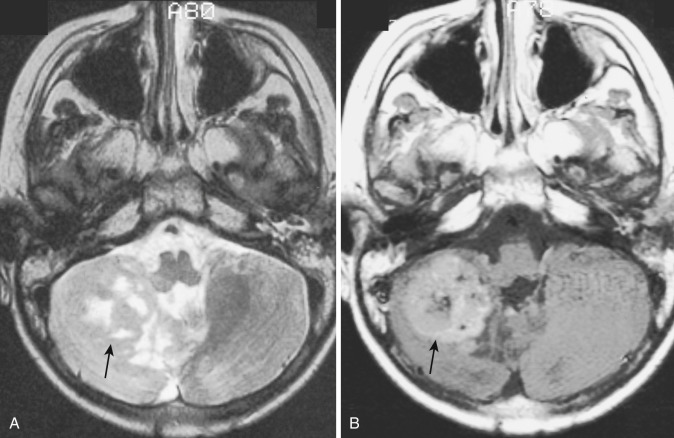
CNS primitive neuroectodermal tumor.
Some 15% of all PNETs of the CNS occur above the tentorium. However, they represent less than 1% of all pediatric brain tumors. Like their infratentorial counterparts, they occur mainly in the very young (65% of patients < 5 years; 85% < 10 years). Patients may present clinically with seizures or with symptoms and signs of increased intracranial pressure. The distribution of these aggressive tumors within the cerebral hemispheres is, in diminishing order of frequency, frontal, parietal, temporal, and occipital. Approximately one third are pineoblastomas; the remainder are central neuroblastomas, ependymoblastomas, ganglioneuroblastomas, medulloepitheliomas, and other rare immature round cell tumors.
On CT, CNS PNETs appear heterogeneous and well circumscribed. The heterogeneity reflects cystic or necrotic changes and intratumoral hemorrhage. The solid elements are hyperdense because of their high cellularity and demonstrate contrast enhancement. Coarse calcifications are described in 50% to 70%, a much higher proportion than in cerebellar medulloblastomas. CNS PNETs share with their infratentorial counterparts a strong propensity for spread via CSF.
MRI also demonstrates heterogeneity of signal intensity on both T1- and T2-weighted images ( Fig. 9-33 ; see Fig. 9-73 ). These are often large tumors with relatively lesser degrees of surrounding edema. The differential diagnosis includes other large well-circumscribed supratentorial tumors such as ependymomas and oligodendrogliomas. As on CT, the solid portions of the tumor demonstrate strong contrast enhancement, a feature that may aid differentiation.
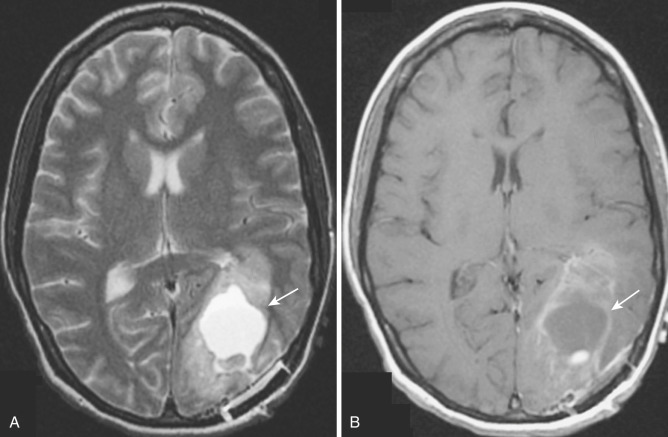
Tumors of the Cranial Nerves
Approximately one third of all intracranial masses are nonglial primary neoplasms of the CNS, almost all of which are extraaxial in origin and location. The most common of these are schwannomas of the cranial nerves and meningiomas.
Within the brain and spinal cord, the nerve fiber tracts are surrounded by oligodendrocytes. However, as the cranial nerves emerge from the brain, there is a transition zone beyond which the oligodendrocytes do not extend but are instead replaced by Schwann cells (also called neurilemmal cells ), which surround and ensheath the extraaxial portions of the cranial nerves, spinal nerve roots and nerves, and peripheral nerves. Within the cranial vault, nearly all tumors arising from these nerves represent schwannomas.
The other major primary tumor of the cranial nerves is neurofibroma. Neurofibromas differ from schwannomas not only in site of origin but also in histology and natural history. Malignant degeneration is essentially unknown in schwannomas, but both malignant degeneration of neurofibromas and primary malignant peripheral nerve sheath tumors are recognized but uncommon occurrences.
Schwannoma.
Schwannoma is a benign (WHO grade I) encapsulated tumor that arises from Schwann cells of nerve sheaths of cranial and spinal nerves. It is estimated to account for between 5% and 10% of primary intracranial neoplasms and for nearly 30% of intraspinal tumors.
Intracranial schwannomas occur in all age groups, but the peak incidence is in the fourth through seventh decades of life. There is a distinct sex predilection, with a female-to-male ratio of 2 : 1, although no such predilection exists in regard to spinal schwannomas.
By far the most common site of intracranial involvement is the superior vestibular division of the eighth cranial nerve. Trigeminal nerve schwannomas are much less common, with facial, trochlear, and abducens nerve tumors only rarely reported. Only the first (olfactory) and second (optic) cranial nerves lack Schwann cell sheaths and are therefore not potential sites of origin. The clinical presentation varies according to the site of origin. Vestibular schwannoma (also called acoustic neuroma or neurinoma ) typically presents with symptoms of a mass in the cerebellopontine angle, including tinnitus, sensorineural hearing loss, and facial paresthesias. Despite the tumor’s origin from the vestibular nerve, symptoms and signs of vestibular dysfunction do not become manifest until relatively late. These tumors have a distinct propensity to involve the sensory nerve roots; motor symptoms are uncommon.
Multiplicity of intracranial schwannomas strongly suggests the presence of neurofibromatosis type 2 (NF2), a congenital inherited disorder associated with a mutation on the long arm of chromosome 22. The occurrence of bilateral vestibular schwannomas (see Fig. 9-35 ) is pathognomonic of this disorder, which has also been termed MISME (multiple inherited schwannomas, meningiomas, and ependymomas). In fact, labeling this disorder “neurofibromatosis” is a misnomer because neurofibromas are not a part of its constellation of abnormalities. Patients with this disorder typically present clinically in the second and third decades of life, much earlier than those with sporadic intracranial schwannoma.
On gross inspection, schwannomas appear as focal well-circumscribed globular or ovoid masses that do not infiltrate the nerve but rather arise and grow eccentrically, displacing the uninvolved portion of the cranial nerve of origin to the side. As the tumor grows and matures, it may undergo cystic degeneration or develop patchy areas of lipid accumulation. Vestibular schwannomas typically enlarge within the cerebellopontine angle cistern and may displace and compress the adjacent pons, medulla, and fourth ventricle (see Fig. 9-21 ). They can attain considerable size before becoming clinically evident. Obstruction of the ipsilateral cerebellopontine angle cistern may occur with formation of one or more extratumoral arachnoid cysts. Trigeminal schwannomas tend to occur more commonly in the parasellar region of the middle cranial fossa than in the posterior fossa, although approximately 25% extend through the tentorial notch, involving both fossae.
Microscopic evaluation of a schwannoma typically demonstrates a well-encapsulated tumor containing collections of spindle-shaped Schwann cells in both compact cellular (Antoni A) and less cellular and more loosely textured (Antoni B) arrangements. Histologic variations are commonly encountered in larger tumors, including intratumoral clusters of lipid-laden cells, confluent microcysts and larger cysts, and dilated vascular sinusoids surrounded by foci of hemorrhage.
The diagnosis of vestibular schwannoma on CT rests on demonstration of a well-circumscribed globular or ovoid, hypodense to isodense (with respect to the adjacent pons and cerebellum) extraaxial mass in the cerebellopontine angle cistern, with its base on the posterior aspect of the petrous temporal bone in the region of the internal auditory meatus. Images displayed in a bone window may demonstrate fusiform widening of the internal auditory canal with erosion of its bony margins. However, soft tissue detail within the cisternal portion of the tumor is often obscured by beam-hardening artifacts caused by the adjacent dense cortical bone and pneumatized air cells of the petrous pyramid. Tumors up to about 1 cm in diameter typically demonstrate homogeneous density and homogeneously strong contrast enhancement, but larger tumors often demonstrate heterogeneity of both density and contrast enhancement because of the presence of intratumoral cystic degeneration, xanthomatous change, or intermixed areas of hypercellularity and hypocellularity.
On MRI, smaller tumors can be more reliably detected, especially within the internal auditory canal, because of the lack of bone-induced artifact and the multidimensional capability of this modality. The transition zone between the central (oligodendroglial) and peripheral (Schwann cell) portions of the eighth (vestibulocochlear) nerve, the presumed site of origin of schwannomas of this nerve, occurs within the canal. Tumors less than 5 mm in diameter can be reliably identified on thin-section T1-weighted MRI, appearing as homogeneously mildly hypointense or isointense (to adjacent brain) ovoid or tubular intracanalicular masses with intense homogeneous contrast enhancement ( Fig. 9-34 ). On T2-weighted pulsing sequences, small vestibular schwannomas appear mildly to markedly hyperintense and may be obscured by the similarity in signal intensity to that of the surrounding CSF.
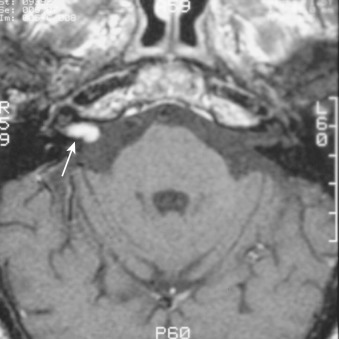
As these tumors enlarge, the intratumoral degenerative changes previously described cause increasing heterogeneity of signal within the main tumor mass in the cerebellopontine angle cistern. On axial images the tumor often has a comma-like shape with a globular cisternal mass medially and a short tapered fusiform extension laterally into the internal auditory canal. Contrast enhancement is seen in nearly all schwannomas and may be homogeneous (two thirds of cases) or heterogeneous ( Fig. 9-35 ; see Figs. 9-21 and 9-34 ). Larger tumors compress and displace the adjacent anterolateral margin of the cerebellar hemisphere posteromedially; displace, compress, and rotate the pons and medulla; and narrow the fourth ventricle, elongating it in the anteroposterior direction (see Figs. 9-21 and 9-35 ). Peritumoral vasogenic edema is frequently observed, and (as noted previously) associated arachnoid cysts are visualized in 5% to 10% of cases.
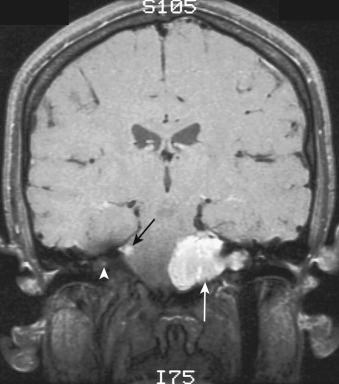
Vestibular schwannomas represent 80% to 85% of cerebellopontine angle masses. The major differential diagnostic considerations include meningioma and epidermoid tumor. Meningioma involving the dura of the posterior margin of the petrous temporal bone can project posteriorly into the cerebellopontine angle and simulate a vestibular schwannoma ( Fig. 9-36 ). Such lesions represent about 10% of cerebellopontine angle masses. Although meningiomas are typically more dense on CT, these two neoplasms may have identical signal intensity characteristics on MRI. Intense homogeneous or slightly heterogeneous contrast enhancement is another shared characteristic (see Fig. 9-36B ). However, meningiomas are usually situated eccentric to the internal auditory canal and form a more obtuse angle with the petrous ridge than schwannomas. Extension of meningioma into the internal auditory canal is uncommon but not unknown, and rarely schwannoma may simulate the appearance of a dural tail on postcontrast images.
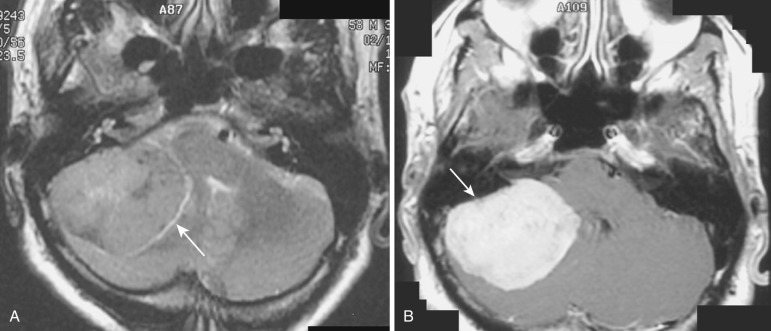
Epidermoid may resemble vestibular schwannoma on noncontrast CT but typically appears more lobulated and more hypodense and tends to insinuate more extensively into the subarachnoid spaces adjacent to the pons without causing major compression of the pons or fourth ventricle. On both CT and MRI, epidermoid does not demonstrate contrast enhancement. On MRI, larger epidermoids appear more homogeneous than schwannomas of similar size and may mimic CSF in intensity with high T2 signal and low T1 signal ; differentiation from arachnoid cyst is made by the demonstration on DWI of restricted diffusion in epidermoid.
Neurofibroma.
Neurofibromas are well-demarcated infiltrative intraneural or diffusely infiltrative extraneural benign tumors that consist of a mixture of neoplastic Schwann cells, perineurial cells, and fibroblasts in a collagenous and mucoid matrix. They are histologically benign and considered WHO grade I. With only extremely rare exceptions, neurofibromas do not arise within the intracranial portions of the cranial nerves. However, they can arise peripherally and extend retrogradely into the cranial vault, notably in the facial nerve (CN VII) and the ophthalmic division of the trigeminal nerve (CN V1).
The tumors exhibiting this retrograde intracranial extension are usually plexiform neurofibromas that involve multiple nerve fascicles or trunks with fusiform multinodular enlargement often described as ropelike. On noncontrast CT, they appear isodense to adjacent muscle, and on MRI they are isointense to adjacent muscle on T1-weighted images and hyperintense on T2-weighted images. Contrast enhancement is variable, with areas of moderate to strong enhancement after IV administration of gadolinium.
Multiple neurofibromas are typically associated with NF1 (von Recklinghausen’s disease), an autosomal dominant congenital disorder associated with a gene defect on the long arm of chromosome 17. Approximately 50% of patients report no family history and are presumed to have new spontaneous germline mutations. In addition to multiple neurofibromas of the spinal and peripheral nerves, NF1 also includes optic nerve and chiasm gliomas (see earlier); foci of “hamartomatous” change of uncertain etiology in the region of the basal ganglia and in the optic tracts and radiations, cerebral peduncles, and brainstem; astrocytomas of the spinal cord; multiple café au lait spots; axillary and inguinal freckling; malignant peripheral nerve sheath tumors; iris hamartomas (Lisch nodules); and dysplastic osseous lesions (sphenoid wing dysplasia, pencil-like hypoplasia of the metaphyses of long bones). Patients with NF1 usually present clinically very early in life, whereas those with sporadic solitary neurofibroma involving a cranial nerve may present in later youth and adult life.
On noncontrast CT, plexiform neurofibroma appears as a poorly circumscribed irregular widening of the involved nerve root, which is isodense with adjacent muscles. Erosion of adjacent bone with widening of bony foramina may be evident. MRI demonstrates the widened nerve, which is isointense to adjacent muscle on T1-weighted images and hyperintense on T2-weighted images. Contrast enhancement after IV administration of gadolinium is the rule, varying from moderate to intense.
Malignant Peripheral Nerve Sheath Tumors.
Malignant peripheral nerve sheath tumors are rare tumors that appear to arise de novo in the cranial nerves, the fifth cranial nerve being more frequently involved than any other. Elsewhere in the body, almost two thirds of reported cases of malignant peripheral nerve sheath tumors have arisen from preexisting neurofibromas, often in the setting of NF1. Approximately 50% occur in association with NF1. The incidence of malignant progression in patients with plexiform neurofibroma is approximately 5%. Imaging findings include irregularity and loss of clear tumor margins together with inhomogeneous contrast enhancement. Evidence of diffuse invasion of the skull base may also be present.
Tumors of the Meninges
A wide variety of primary tumors can arise from and develop within the meninges. By far the most common is meningioma. Additionally, nonmeningothelial mesenchymal tumors—both benign (lipoma, chondroma, benign fibrous histiocytoma) and malignant (chondrosarcoma, osteosarcoma)—can originate in the meninges. Also, tumors of melanocytic origin can in rare cases arise within the intracranial meninges. Finally, although the histogenesis is uncertain, neuropathologists usually assign hemangioblastomas to this category.
Meningioma.
Meningiomas are solid, well-circumscribed, highly cellular, slow-growing tumors that are usually benign histologically (WHO grade I). These sometimes spherical, sometimes lobulated, and sometimes flat and broad-based tumors are composed of neoplastic meningothelial cells originating from the arachnoid layer of the meninges, with a broad attachment to the adjacent dura. Most commonly they project inward from the dura, indenting and compressing the underlying brain, causing neurologic symptoms and signs through compression of the adjacent cortex.
Meningiomas are the most common primary extracerebral tumors of the CNS, accounting for approximately 20% of primary brain tumors. They occur mainly in middle and old age, with a peak incidence in the fifth through seventh decades of life; these tumors can, however, be found in all age groups. Meningiomas exhibit a strong sex predilection, occurring preponderantly in females, with a female-to-male ratio of at least 2 : 1. However, meningiomas associated with hereditary tumor syndromes such as NF2 generally occur in younger patients and do not demonstrate a gender predilection.
Common sites of origin are in the frontal and parietal convexities and the parasagittal region (in close association with the falx cerebri); tumors in these locations constitute about 50% of all intracranial meningiomas. Meningiomas arising in relation to the sphenoid wings, olfactory grooves, sylvian fissures, and parasellar regions represent about 35%. Less than 10% arise below the level of the tentorium, mainly from the meninges overlying the clivus and petrous pyramid and the leaves and free edge of the tentorium. Multiple meningiomas are reported in 6% to 9% of cases, occurring as a component of NF2 and less commonly sporadically.
Although most meningiomas are slow-growing and well-encapsulated globular masses, variations in shape and histology are not unusual. Invasion of the dura and encasement or invasion of the nearby dural venous sinuses are common. Tumors arising in relation to the sphenoid wing are frequently flat (en plaque) and tend to invade through both layers of the cranial dura into the adjacent bone, provoking a notable bony reaction with thickening and sclerosis (meningioma bone). Such bony changes are a source of considerable controversy; many clinical neuroscientists regard bony sclerosis and thickening as highly indicative of tumor infiltration into the haversian canals of the calvaria, whereas others state that such hyperostosis can also occur without histologic evidence of bone infiltration by tumor. Meningiomas arising adjacent to the cribriform plate and planum sphenoidale and those occurring over the high cerebral convexities are more typically rounded and invaginate the underlying brain; however, they can also show a propensity for stimulating adjacent bony thickening and sclerosis ( Fig. 9-37 ).