Fig. 1
2D representation of the CD20 antigen (previously known as “B1”). Note that antibodies used in RIT bind to the extracellular domain. This figure was published in American Journal of Pathology, 136, Mason et al. (1990). Figure 1, page 1217, Copyright Elsevier
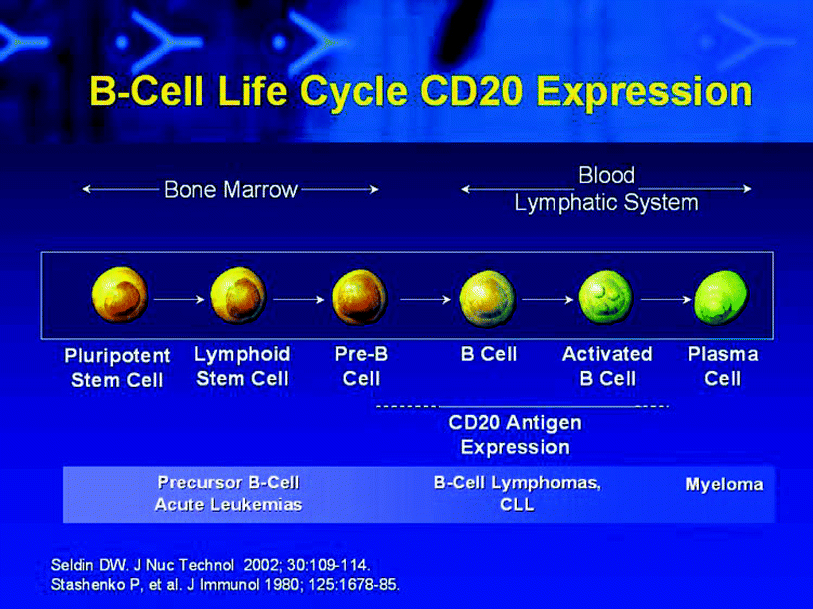
Fig. 2
Schematic of B-cell maturation pathway from pluripotent hematopoietic stem cell (left-most) to activated B-cells, with loss of much of the CD20 expression on plasma cells. CD20 is expressed on B-cell lymphomas. This research was originally published in JNMT (Seldin 2002) Figure 1. © by the Society of Nuclear Medicine and Molecular Imaging, Inc
Two functional types of anti-CD20 antibodies have been described, type I (rituximab-like) and type 2 (tositumomab-like). While it was originally thought that antibody bound to CD20 did not internalize substantially, it has been more recently shown that in live tumor cells the rituximab-like antibodies internalize more rapidly than the tositumomab-like antibodies. This may have practical implications for radioimmunotherapy since most radioiodinated antibodies, when internalized, generally lose their radiolabel resulting in reduced tumor radiation dose. The relative lack of internalization of tositumomab-like type 2 antibodies is an important consideration and a favorable one for 131I-tositumomab therapies. There remains some controversy related to the extent of CD20 internalization following antibody binding, although it is typically less than that of many other antigenic targets (Johnson and Glennie 2003).
The effects of radiolabeled antibodies must be considered in the context of the incremental anti-tumor activity above that delivered by the unlabeled antibody alone. Currently, there are several fully humanized anti-CD20 monoclonal antibodies such as ofatumomab (type 1 antibody), as well as the well-known mouse-human chimeric monoclonal antibody rituximab (type 1 antibody). However, the current US FDA-approved radioimmunotherapies were developed from and based on first generation murine monoclonal antibodies.
At the time of preparation of this chapter, two radiolabeled anti-CD20 monoclonal antibodies,Yttrium-90 (90Y)-ibritumomab tiuxetan and Iodine-131 (131I)-tositumomab are currently approved by the US Food and Drug Administration for the treatment of NHL as part of the Zevalin® (Spectrum Pharmaceuticals, Irvine, CA) and Bexxar® (GlaxoSmithKline) therapeutic regimens, respectively.
90Y-ibritumomab tiuxetan is composed of the radioisotope 90Y and the murine monoclonal antibody ibritumomab and is discussed elsewhere in this textbook (Zevalin package insert 2009). 131I-tositumomab is composed of the 131I radioisotope and the murine monoclonal antibody tositumoma (Bexxar package insert 2003) 15. Both the unlabeled antibody and the radioactivity contribute to the drugs’ mechanism of action.
Tositumomab, the monoclonal antibody on which Bexxar is based, recognizes the type II epitope of the extracellular domain of the CD20 antigen. This epitope has received considerable attention as there is evidence that type II antibodies are up to 5 times more potent than type I (rituximab-like) antibodies in depleting human CD20 B-cells, in large part due to low internalization rates of the type II tositumomab (Beers et al. 2010). Of note is that antigenic modulation is particularly slow in follicular lymphomas, the histology in which RIT has been most effective.
As stated earlier, CD20 is found on the surface of pre-B lymphocytes, mature B-lymphocytes, and >90 % of B-cell NHLs (Anderson et al. 1984; Tedder et al. 1985; Cardarelli et al. 2002). Formation of the antibody-antigen complex results in induction of apoptosis, complement-dependent cytotoxicity, and antibody-dependent cytotoxicity (Boross and Leusen 2012). The radioisotopes emit beta radiations which deposit enough energy in the tumor to result in cell death. Because the distribution of radiolabeled antibody within a tumor can be heterogeneous after iv delivery (Brown et al. 1997), an additional advantage of RIT is that the beta-emissions of the radiolabel can also target the surrounding malignant cells via the cross-fire effect. This probably results in the killing of residual microscopic disease in cells that the unlabeled antibody did not reach either by direct lethality of the radiation or potentially by affecting the tumor blood supply.
1.2 Development
131I-tositumomab (originally called the “B-1” or “anti B-1” antibody) plus unlabeled tositumomab therapy of lymphoma was developed in a somewhat unconventional manner. The CD20 antigen, originally known as the B-1 antigen, was among the first pan B-cell marker antigen/antibody pairs (Stashenko et al. 1980; Tedder et al. 1985). A challenge with the CD20 target is that CD20 is present on normal B-cells as well as B-cell lymphomas (Fig. 1). However, an advantage of CD20 is that it is not present on pluripotent hematopoietic stem cells (Fig. 2). Pre-clinical studies showed that the I-131 anti-CD20 antibody,131I tositumomab, had greater anti-tumor effects than unlabeled tositumomab (Buchsbaum et al. 1992).
To deal with the dual challenges of circulating normal B-cells expressing the CD20 antigen, which would be the first cells to interact with the infused antibody, and the expected variability of clearance, a method was developed for patient dosing which addressed both challenges. The challenge with expected variability in clearance is that varying mCi doses would need to be infused to deliver the same radiation-absorbed dose (Fig. 3). Only by knowing if patients are fast or slow clearers of the radioantibody could the proper mCi amount of activity to infuse be determined.
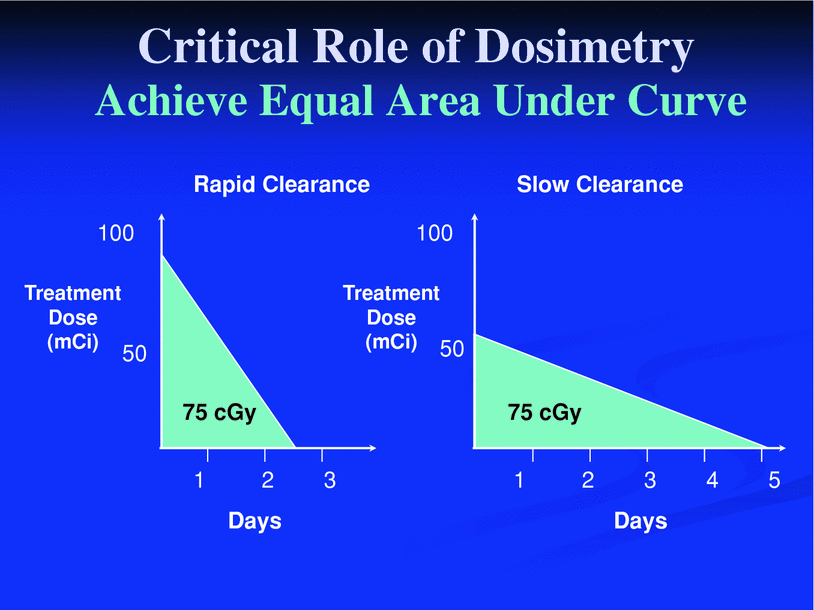
Fig. 3
Schematic example of dosimetric approach to 131I-tositumomab therapy. Patients have variability in clearance of radioactivity from the body. Those with fast clearance will require more administered radioactive molecules (left) than those with rapid clearance (right) to deliver the same cumulative activity (dose) levels. Reprinted with permission from Wahl (2005). Figure 3A
In the phase I study, varying masses of unlabeled antibody pre-dosing were evaluated in an intra-patient comparison, coupled with individualized patient and tumor dosimetry. Thus, a tracer dose of radioantibody, the “dosimetric dose,” was given, and then daily gamma camera/or probe counts were obtained to determine the rate of clearance of the radioactivity from the patient (Kaminski et al. 1993).
In individual patients, varying masses of unlabeled monoclonal antibody were given before up to three separate dosimetric doses, up to three different mass levels (including no unlabeled antibody pre-dose). A simplifying assumption was made that the radioantibody was distributed rather uniformly in the patient, and a dosimetric calculation was made to determine the appropriate dose for a therapeutic effect. The dose escalation was performed guided by the projected total body radiation-absorbed dose with the maximum tolerated dose (MTD) being hematopoietic toxicity. An overview of the final Bexxar treatment regimen is shown in Fig. 4.

Fig. 4
Bexxar therapeutic regimen as currently FDA approved. Note that there is thyroid blockade with unlabeled iodine, then administration of unlabeled tositumomab. This is followed by administration of radiolabeled tositumomab for dosimetry purposes. Total body scans are obtained at three time points allowing for determination of the total body clearance rate. This information is used to determine the mCi required to deliver the therapeutic dose. The therapeutic dose is patient-specific, but typically designed to deliver a 65–75 cGy targeted total body dose. This therapeutic dose is preceded by an unlabeled antibody infusion of 450 mg cold tositumomab, over 1 h. Reprinted with permission from Wahl (2005). Figure 4
As shown in Fig. 5, a patient receiving no unlabeled antibody pre-dose on the baseline study and a large mass of unlabeled antibody pre-dose on a follow-up study, had significant effects on the in vivo biodistribution of the radioantibody. Low anti-CD20 mass pre-dose (or no protein mass pre-dose) was associated with rapid clearance of the radioantibody from the blood and a lower tumor radiation absorbed dose than when the radioantibody dosing was preceded by unlabeled anti-CD20 antibody. Higher protein masses were associated with a higher area under the curve (AUC) in the blood (Fig. 6).
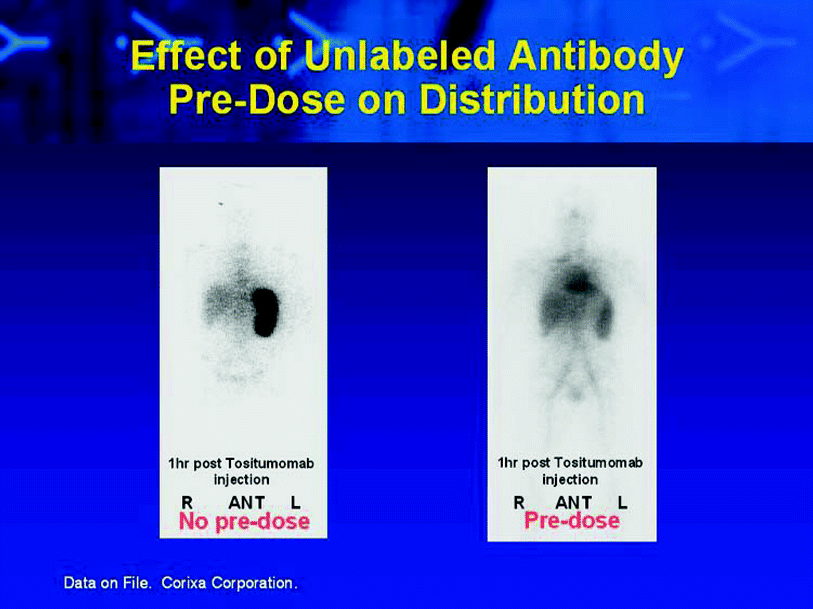
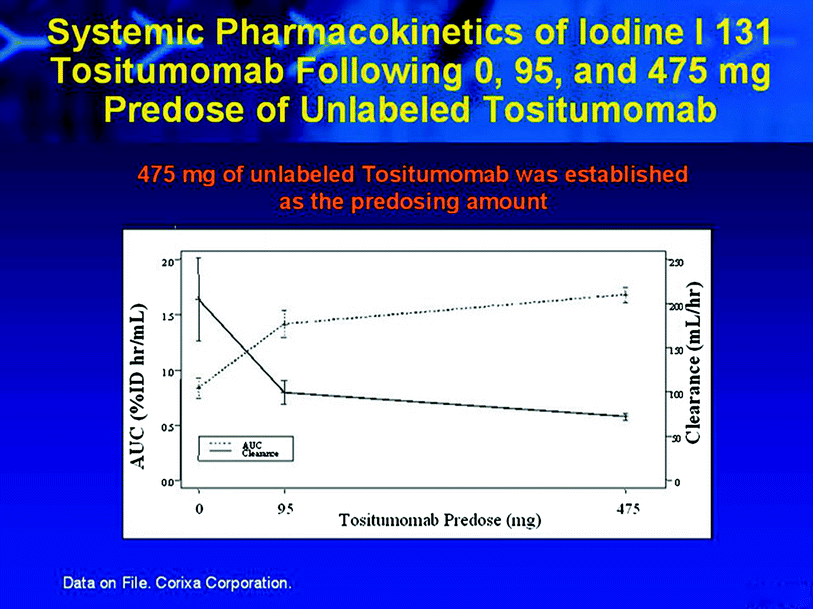
Fig. 5
Example from the phase I study demonstrating a patient who was imaged twice with 131I-tositumomab. Images on the left show intense splenic activity at 1 h post-injection of approximately 15 mg of 131I-tositumomab. Image on the right was obtained about 2 weeks later, in this instance, unlabeled tositumoamb pre-dose was performed before the 131I-tositumomab was given. A much different biodistribution is observed in this case with much less splenic radiotracer uptake and more radioantibody available in the blood pool for delivery to tumor foci. Reprinted with permission from Wahl (2005). Figure 2
Fig. 6
Effect of unlabeled antibody pre-dose of biodistribution of radiolableled anti-CD20 antibody in vivo. The systemic AUC is greater with a substantial antibody pre-dose than if there is no pre-dose of unlabeled antibody. This is likely the case as the labeled antibody in low protein masses binds to the CD20 present on circulating tumor cells and is cleared more rapidly than if the radiolabeled antibody was given after an unlabeled antibody pre-dose, and thus can bypass some of the circulating CD20 positive cells, remaining in the blood for a longer period of time
Tumor dosimetry in this phase I study showed no decrement in tumor targeting relative to the total body-predicted radiation-absorbed dose at higher unlabeled tositumomab infusion mass levels. While the total body clearance of the dosimetric dose was slower at higher protein mass levels, there was no clear blocking of tumor targeting of the radioantibody.
Higher mass doses of tositumomab by itself were expected to have higher anti-tumor activity due to the known immunological effects of the unlabeled antibody (Buchsbaum et al. 1992). Thus, the protein mass for the therapeutic regimen was selected to be 450 mg, the highest mass used in the phase I study. It is notable, however, that this determination was made in patients who had not received prior rituximab therapy and who had rather substantial tumor burdens.
It is possible that the settled up, 450 mg, unlabeled anti-CD20 pre-dose could be too high for patients with low tumor burdens or who have had recent rituximab therapy. It is clear by basic immunological principles that unlabeled antibodies will ultimately be able to block the targeting of radiolabeled antibodies to tumor. The pre-dose of unlabeled tositumomab effectively blocked the uptake of the dosimetric dose of 131I-tositumomab to circulating B-Cells and to the spleen and there has been some evidence of tumor blocking in animal models (Gopal et al. 2008).
The other challenge in using an antibody which cross reacts with normal tissues was that the biodistribution might not be consistent depending on the amount of antibody given, the mass of tumor cells, and the number of CD20 positive cells accessible from the blood. The phase I dose escalation was performed based on patient-specific dosimetry, in which the total body radiation dose was increased in 10 cGy increments, as predicted by the dosimetric dose. The total mCi administered was driven by patient-specific dosimetry and not by mCi/kg or simply mCi. This showed that the maximum tolerated dose (MTD) was approximately 75 mCi in heavily pretreated patients who had not received rituximab therapy (Fig. 7). In the early report of the phase I study, safety was encouraging as was anti-tumor response, with 6/9 treated patients responding and four having complete responses, which was supported as the study was expanded to MTD (Kaminski et al. 1993, 2000; Wahl et al. 1998b).
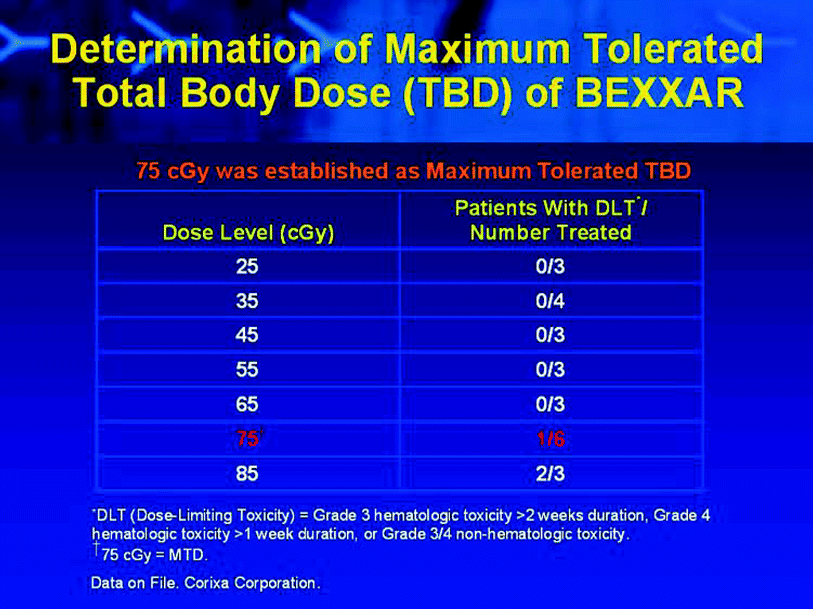
Fig. 7
Data showing how dose escalation was performed, lowest therapeutic dose was 25 cGy, which was gradually increased to 85 cGy in a 3 + 3 escalation. In this group of heavily pretreated patients, the Maximum Tolerated Dose (MTD) was identified as 75 cGy
There is a great deal of discussion regarding personalized cancer therapies and the use of imaging to guide therapies. 131I-tositumomab is a personalized therapy on at least two levels: (1) the molecular target, CD20, must specifically be present on the tumor for the treatment to be used and, (2) the administered dose is individualized based on the patient-specific dosimetric imaging.
1.3 Methodological Considerations
The dosimetry and radiation safety aspects of 131I-tositumomab + unlabeled tositumomab therapy have been discussed in detail elsewhere (Gates et al. 1998). In the US, under current NRC rules, the therapeutic regimen can be given in the outpatient setting in nearly all cases, with close attention to radiation safety (Gates et al. 1998). It is important to note that clinical trial data from several studies have shown there to be considerable variability in the total body clearance rates of tracer doses of 131I-tositumomab. This results in considerable variability (when coupled with differences in body habitus) in the mCi required to deliver the targeted 75 cGy total body dose that is appropriate in most NHL patients with platelet counts >150,000 mm3 (Fig. 8).
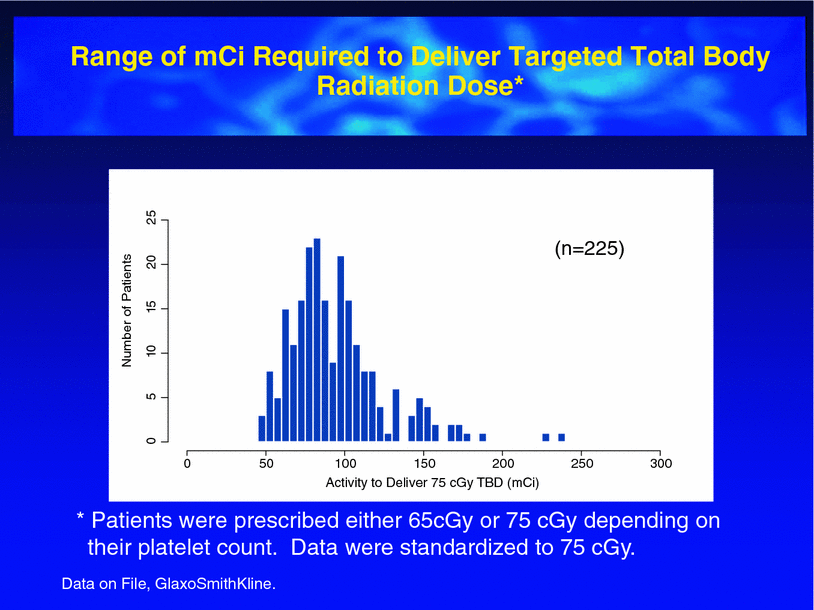
Fig. 8
Range of mCi required to deliver 75 cGy TBD to 225 patients treated with 131I-tositumomab. Note the considerable variability, which is accounted for by the personalized dosimetry approach
In the FDA-approved version of the drug (see clinical data below), a simplified dosimetry scheme in which a limited number of data points are used has been employed (Fig. 4). In brief, the clearance of 131I-tostitumomab from the whole body is monoexponential, in general, and the clearance can be represented well by just 3 time points of imaging (or probe counts). We prefer gamma camera imaging to secure information on biodistribution, but early clinical trials were done and included a whole body NaI probe system focused at the central body, securing kinetic data.
By knowing the patient’s weight, by determining a maximum effective mass (to minimize overdosing of obese patients based on weight), it was possible to develop tables of activity-hours (mCi-hrs) required to deliver a given radiation dose to a patient. These values are predicted by the dosimetry scans. The activity-hours to deliver 75 cGy are noted in Fig. 9. A very simple calculation then follows to determine the therapeutic mCi dose for a specified total body dose level (Fig. 10). The FDA-approved drug, Bexxar, depends on use of this dosimetry approach, which has been discussed in several reports (Kaminski et al. 1992, 1993; Wahl et al. 1998a). This dosimetry approach also works well for 131I-rituximab therapy, which has been deployed in Australia (Calais and Turner 2012).
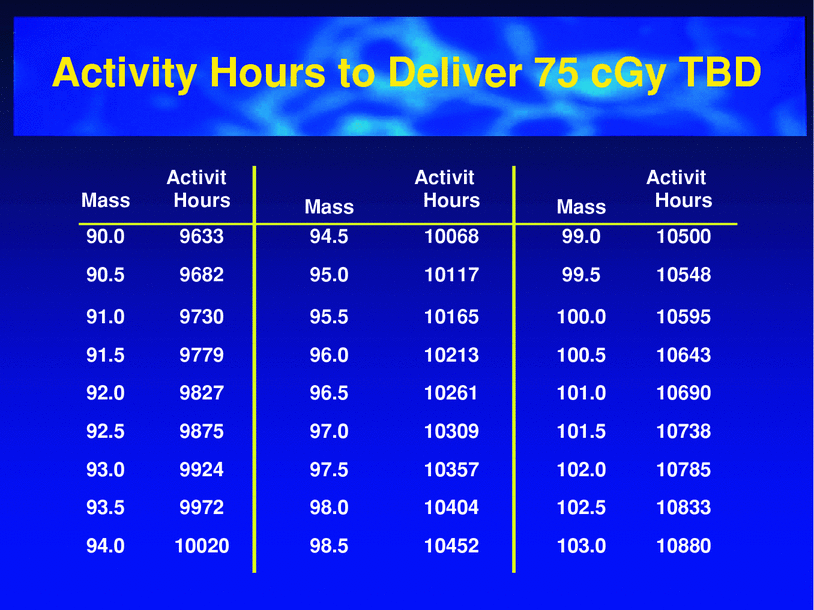

Fig. 9
Tabular data indicating the activity-hours required to deliver 75 cGy TBD. These calculations are based on the patient mass and an assumed uniform distribution of radiotracer through the patient’s whole body. (Bexxar package insert 2003)
Fig. 10
Simplified dosimetry to calculate the therapeutic dose of 131I-tositumomab. The activity hours determined for each patient, based on their body mass, is divided by the residence time (determined from the three whole body imaging time points). This multiplied by the desired total body dose/75 cGy (most commonly = 1) gives the therapeutic mCi required in the specific patient. (Bexxar package insert 2003)
1.4 Efficacy and Tolerance of RIT in Recurrent NHL
In clinical trials investigating 131I-tositumomab in patients with recurrent NHL, including both follicular recurrences, or transformed lymphomas, overall response rates have ranged from 60–83 %, with complete response (CR) rates of 15–52 %. The median duration of response with RIT ranges from 6–16 months for all responders, but seems to be associated with the quality of the initial response. At the time of publication of several studies, the median duration of response for patients who achieved a CR was not yet reached. It was very clear from the early studies that achieving a CR resulted in a high probability of long-term disease free status (Kaminski et al. 2000, 2001); Vose et al. 2000).
Higher response rates have been reported for RIT compared to the unlabeled antibody. In a cross-over study of 131I-tositumomab, 42 % of patients nonresponsive to unlabeled tositumomab achieved a CR after receiving a therapeutic dose of 131I-tositumomab (Davis et al. 2004). This supports the value of the radioactive component of the therapy, though it also shows there is anti-tumor activity for the unlabeled tositumomab. A partial explanation for the improved overall response rates with the radioactive monoclonal antibodies is that the addition of the radioactivity probably allows more cells to be targeted and killed due to the radiation cross-fire effect.
Although the initial results of the studies investigating 131I-tositumomab in recurrent NLH (mostly follicular) were excellent, the studies preceded those of rituximab. These trials included mostly patients who were rituximab naϊve and studies were needed to show efficacy of RIT in the group of patients previously treated with rituximab, which is now a routine component of B-cell lymphoma regimens.
Horning et al. described the therapeutic efficacy of whole body doses of 65–75 cGy of 131I-tositumomab (Bexxar) in 40 patients (24 rituximab nonresponders, 11 < 6 month responders, and 5 > 6 month responders) (Horning et al. 2005). In addition, the population included 59 % who were chemotherapy resistant. This group of patients had been extensively pretreated, with a median of four prior types of treatment before radioimmunotherapy. The overall response rate in this difficult to treat group was 65 %, with a CR rate of 38 %. Responses were not related to the prior response to rituximab. The median PFS was 24.5 months for responders and was not yet reached for those patients achieving a CR—again supporting the durability of a CR in response to the Bexxar regimen. Of particular note was an 86 % response rate (57 % CR) in patients with low-grade follicular lymphomas and tumors <7 cm in diameter. Toxicities were typically hematological, with grade 3 or 4 marrow toxicity in 50 % of cases. These strong data were instrumental in the FDA approval of the drug and its labeled indication (Horning et al. 2005).
Numerous prognostic factors have been evaluated in an attempt to identify which patients are most likely to respond to RIT. In individual studies, factors reported to be associated with response are low-grade follicular histology, nonbulky disease, no bone marrow involvement, fewer prior regimens, normal serum lactate dehydrogenase levels, a lower fractional prognostic index (IPI), a higher total body radiation dose, higher tumor radiation dose, and no prior history of transplants (Kaminski et al. 2000, 2001; Vose et al. 2000; Davis et al. 2004; Horning et al. 2005). Of all of these, low-grade follicular histology and nonbulky disease at the time of treatment are the most reliable predictors, as was well shown in the study by Horning et al. (2005).
In patients with NHL, RIT is generally well tolerated. The most common adverse events are serious cytopenias and these are experienced by a majority of patients. 60–70 % of patients enrolled in clinical trials developed grade III/IV thrombocytopenia and/or neutropenia. Count nadirs typically occurred between 4–7 weeks post-RIT and cytopenias may last for approximately 30 days. Consequently, monitoring of complete blood counts is required for at least 10–12 weeks post-therapy. Hematologic support is required for ∼15–32 % of patients after RIT (Kaminski et al. 2000, 2001; Vose et al. 2000; Davis et al. 2004; Horning et al. 2005).
Additional nonhematologic short-term side effects are usually mild and may include fatigue, asthenia, nausea, and fever. There are several long-term side effects that prescribing nuclear medicine physicians/radiologists, oncologists, and patients must also be aware (Kaminski et al. 2000, 2001; Vose et al. 2000; Davis et al. 2004; Horning et al. 2005). In patients with recurrent NHL treated with RIT, human anti-mouse antibodies (HAMA) are found in 4–11 %. Acute side effects due to HAMA (such as serum sickness-like illness) are not typically seen in this population due to a history of prior therapies and relative immune compromise, but could affect future treatment decisions (treatment with additional monoclonal antibody, eligibility for clinical trials) or interfere with certain laboratory evaluations. HAMA is far more common if 131I-tositumomab is used as the initial therapy of NHL.
Hypothyroidism has been reported to occur in ∼18 % of patients who receive 131I-tositumomab despite thyroid blockade prior to therapy, but is probably less with careful attention to thyroid blockade. Secondary myelodysplastic syndrome/acute myelogenous leukemia (MDS/AML) has been reported in 2–4 % of patients with recurrent NHL who received RIT (Zevalin package insert 2009; Bexxar package insert 2003). The patients in these studies were heavily pretreated, and the role of RIT versus exposure to prior cytotoxic agents and a history of NHL in the development of MDS/AML are unresolved.
Czuczman et al. (2007) retrospectively reviewed the data from 746 patients previously treated with 90Y-ibritumomab tiuxetan from 1996 to 2002 who were participating in clinical trials and compassionate use trials. Nineteen patients (2.5 %) developed MDS/AML at a median of 5.6 years (1.4–13.9 years) after RIT. The annualized incidences of MDS/AML after NHL diagnosis (0.3 % per year) and after treatment (0.7 % per year) were not more than those expected in heavily pretreated patients with NHL. This experience may be relevant to the experience with 131I-tositumomab. At a median follow-up of 5.1 years, no cases of MDS/AML were reported in 76 patients with untreated follicular NHL treated with 131I-tositumomab as front-line therapy, though a single case has been reported after longer term follow-up (Kaminski et al. 2005). Patients with NHL are generally at higher risk for the development of MDS/AML and should be monitored carefully.
1.5 Front-line and Consolidation Therapy with RIT
RIT has also been investigated in front-line and consolidation settings. In both instances, single doses of RIT are effective and can produce long-term remissions. In 76 untreated patients with follicular NHL, Kaminski et al. reported a 95 % overall response rate and a 75 % complete response rate to a single dose of 131I-tositumomab (Kaminski et al. 2005). The estimated median progression-free survival rate at 5 years was 59 % and the mean progression-free survival was 6.1 years. The relapse rate decreased progressively with time from the first RIT and suggests that the quality of initial response is important for subsequent relapse.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


