Model
Model establishment
Advantages
Disadvantages
STZ
Wei et al. (2003)
Type 1: chemical toxicity of STZ to target insulin-producing β cells
Inexpensive; can be established in different strains; easy to breed
Toxicity & off-target effects of STZ; degree of renal injury is strain dependent
Akita
Yoshioka et al. (1997)
Type 1: mutation in Ins2 gene resulting in pancreatic β cell failure
Autosomal dominant trait; stable insulin resistant diabetes phenotype; diabetes onset is not through systemic immunogenic alteration
Modest renal injury only available in C57BL/6 strain
NOD
Anderson and Bluestone (2005)
Type 1: autoimmune destruction of pancreatic islets
Commercially available; spontaneous defective β cells
Ketoacidocis is mild; females are more susceptible to the diabetic phenotype
db/db
Chen et al. (1996)
Type II: mutation in leptin receptor gene
Widely used
Predictable elevation in albuminuria and mesangial expansion
Autosomal recessive trait; homozygotes are infertile; albuminuria may not be progressive
OVE26
Zheng et al. (2004)
Type I: overexpression of calmodulin gene leading to β cell toxicity
Hyperalbuminuria; High blood pressure & GFR decrease;
Enlarged glomeruli & tubulointerstitial fibrosis. Available on the more sensitive FVB background
Difficult to maintain & high mortality rate; Requirement for FVB background limits breeding of OVE26 mice with KO mice on other backgrounds
Agouti yellow obese (Ay/a)
Yen et al. (1994)
Type II: mutation of the agouti gene resulting in insulin resistance
Available in KK & C57BL/6 strain;
Phenotype in KK background is more pronounced (renal injury with high albuminuria); Intact Leptin signaling pathway
Only males develop overt hyperglycemia; Nephropathy phenotype is less robust in strains other than KK; Susceptible to tumor development
Goto-Kakizaki
Goto et al. (1976)
Type II: decreased pancreatic β cell mass & impaired insulin sensitivity
Insulin deficient & resistant. Thickening of glomerular & tubular basement membranes; Renal lesions, peripheral nerve changes, retinal abnormalities
Moderate hyperglycemia; No progressive proteinuria & glomerulosclerosis; Expensive
Zucker Diabetic FattyFinegood et al. (2001)
Type II: mutation in the Leptin receptor resulting in high circulating Leptin levels
Insulin resistant at young age with progressive hyperglycemia, Defective β cells; Widely studied
Males are more susceptible to developing diabetic phenotype
17.4 Current Therapeutic Strategies and Challenges
Current therapeutic strategies for management of DN are centered on treatment of the known predisposing risk factors: Hypertension, hyperglycemia, dyslipidemia and occurrence of cardiovascular events. Glycemic control, body-weight reduction through bariatric surgery, lipid management etc. have been some of the standard approaches for metabolic control (Fernandez-Fernandez et al. 2014). Although these therapeutic strategies offer renoprotective benefits to slow down the progression of DN, they fail to fully protect against progression to ESRD. We will be addressing some of the primary therapeutic strategies against DN and their associated challenges in the next section.
17.4.1 Glycemic Control
Intensive blood glucose control can decelerate the development of microvascular complications in patients with DN (Gross et al. 2005). In the Kumamoto Study, type 2 diabetic patients receiving multiple insulin therapies for strict glycemic control had slower progression of nephropathy and microvascular complications, including retinopathy (Shichiri et al. 2000). However, the beneficial effects of strict glycemic control are restricted to early-stage DN, prior to the development of albuminuria. Even with strict glycemic control, diabetic patients with developed microalbuminuria do not benefit from a decreased rate of progression to macroabuminuria. (DCCT 1995 and Microalbuminuria Collaborative Study Group 1995).
17.4.2 Renin Angiotensin System (RAS) Blockade
Renin-angiotensin system (RAS) blockade, with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs), produce positive effects on DN-related albuminuria. Both ACE inhibitors and ARBs are not only effective treatment options for hypertension, but also have an additional renoprotective effect independent of blood pressure reduction. This renoprotective effect is associated with diminished intraglomerular pressure and flow of proteins into the proximal tubule (Thurman and Schrier 2003). This renoprotective effect decreases urine albumin excretion (UAE) and progression to more advanced stages of DN. Various clinical studies have shown that ACE inhibitor and ARB therapies can decrease the rate of progression towards overt DN (Parving et al. 2001) and reduce UAE (Viberti and Wheeldon 2002; Andersen et al. 2003). However, in a recent large-scale clinical study involving 1448 type II diabetic patients, increased adverse effects of combined RAS blockade (ACE inhibitors in combination with ARBs) for treatment of DN have been reported (Fried et al. 2013). The study was terminated early due to safety concerns. Similarly, several other clinical trials with PKC inhibitors, ACE, advanced glycation end products (AGEs), aldose reductase etc. have failed to generate positive effects against DN.
17.4.3 Kidney Transplantation
DN is the leading cause of ESRD in the western world (2). For patients with ESRD, the only treatment options available are either dialysis or kidney transplantation (Go et al. 2004). Although dialysis is a lifesaving alternative that can clear out waste and toxins from the blood similar to the kidneys, it is only equivalent to 10 % of healthy kidney function. Besides, long-term dialysis is associated with various complications, such as cardiovascular disease, hernia, peritonitis, etc. (Collins et al. 2009; Diaz-Buxo et al. 2013; Lok and Foley 2013 ; Stuart et al. 2009). The average life expectancy of a patient starting on dialysis is generally 3–5 years (Stokes 2011 and USRDS 2009). The best therapeutic strategy for ESRD patients is kidney transplantation. Kidney transplantation can extend the life expectancy of patients compared to those who stay on dialysis. On average, transplant of a living donor kidney can function for 12–20 years, and a deceased donor kidney from 8 to 12 years (Traynor et al. 2012). Studies have shown that kidney transplantation is especially beneficial for younger patients, but even older adults can favor from an average of 4 or more years of life span compared to patients on dialysis (Briggs 2001; Knoll 2013). However, kidney transplantation is a major surgery with phased recovery. The long-term success of the transplant is limited by the host’s immune response to the foreign kidney graft. Clinical studies have shown that transplants from human leukocyte antigen (HLA)-identical siblings result in highest graft survival (99.17 %, 91.84 %, and 88.96 % at 1, 3, and 5 years, respectively) (Kessaris et al. 2008). To prevent immune rejection, transplanted patients will be required to routinely take immunosuppressant drugs for the rest of their lives. The prolonged immunosuppressant therapies have various adverse effects, including bone disease, hyperglycemia, and most importantly increased susceptibility to opportunistic infections (Alangaden et al. 2006; Marcen 2009). Another major drawback of kidney transplantation, like in any other form of transplantation, is the lack of organ donors. Due to the existing imbalance between demand for kidney transplantation and the number of donor organs, many patients suffering from kidney disease have no option but to remain on dialysis.
17.5 Gene Therapy: Techniques and Vectors – Applications in Diabetic Nephropathy
Many diseases, including DN, result from dysregulation of endogenous genes. Gene therapy aims to overcome deficiencies in endogenous gene expression and regulation, with the goal of restoring normal structure and function. Unlike the short-lived effect of pharmaceutical drug treatments, gene therapy offers a more prolonged cellular protein expression for a sustained therapeutic effect. The success of gene therapy largely depends on the development of vectors or vehicles that can selectively and efficiently deliver genes to targeted cells with minimal toxicity. Broadly, the gene delivery systems can be divided into two categories: Viral and non-viral based.
17.5.1 Viral Gene Delivery
The viral vector delivery system is a popular method of transfection due to its ease of production, having a high functional titer and its ability to infect various cell types (Loiler et al. 2003; Work et al. 2009). A gene of interest can be integrated into a viral vector, and exposure of the virus to the targeted cells can lead to transfection. This leads to expression of the transgene in the host cell, resulting in a desired therapeutic effect. There are several pre-clinical studies using viral vectors for targeting DN (Table 17.2). Although this method has been used for clinical studies of cardiovascular disease (Jessup et al. 2011; Grines et al. 2003), there are as yet no clinical studies of viral gene transfection against DN. Despite being approved for clinical trials, the use of viral vectors has several potential drawbacks, including cytotoxicity, expensive production, high immunogenicity and the risk of mutagenesis in the host genome (Thomas et al. 2003).
Table 17.2
Preclinical studies of gene therapy (non-UMGD) in diabetic nephropathy
Study | Gene | Animal model | Delivery method | Major findings |
|---|---|---|---|---|
Dobrzynski et al. (2002) | Human adrenomedullin (AM) | STZ-induced diabetic rats | iv injection of adenovirus vector | Reduced renal hyperglycemia-induced glycogen accumulation and tubular damage; Increased cAMP & cGMP levels in urine, pAkt & membrane-bound GLUT4 levels in skeletal muscle; Increased body weight & prevented renal dysfunction via Akt signaling pathways |
Dai et al. (2004) | Human hepatocyte growth factor (HGF) | Male STZ-induced diabetic mice | iv injection of naked pDNA | Decreased albuminuria & proteinuria; Improved development of DN by reducing fibronectin & collagen deposition; Prevented glomerular mesangial growth; Inhibited myofibroblast activation & kidney cell apoptosis; Attenuated urinary TGF-β1 protein level |
Kagawa et al. (2006) | Human HGF | C57BL/KsJ-db/db mice | im injection of adenovirus vector | Ameliorated creatinine clearance, renal tubule fibrosis & glomerular sclerosis; Decreased TGF-β1 expression; Reduced glomerular endothelial cell & tubular epithelial cell apoptosis; Improved renal function along with long-term survival |
Zhang et al. (2006) | Translocase of inner mitochondrial membrane 44 (TIM44) | STZ-induced diabetic CD-1 mice | iv injection of hemagglutination virus of Japan-envelope vector | Increased import of antioxidative enzymes into mitochondria; Suppressed cell proliferation & apoptosis; Reduced proteinuria & renal cell hypertrophy |
Yuan et al. (2007) | Human tissue kallikrein (HK) | STZ-induced diabetic rats | iv injection of recombinant adeno-associated virus | Ameliorated clearance of creatinine; Increased urinary osmolarity; Reduced urinary micro-albuminuria; Reduced diabetic renal damage |
Kondo et al. (2008) | Soluble TGF-β2 receptor | STZ-induced diabetic mice | im injection of adenovirus vector | Reduced fibrosis in glomeruli & renal tubules; No effect on glucose metabolism, albumin excretion in urine & renal function |
Ortiz-Munoz et al. (2010) | SOCS-1 & SOCS-3 | STZ-induced diabetic mice | iv (renal vein) injection of adenovirus vector | Improved renal function; Reduced renal lesions; Decreased action of STAT-1 & -3, expression of pro-inflammatory & fibrotic proteins; Inhibited mesangial expansion, fibrosis & influx of macrophages |
Shi et al. (2010) | SOCS-1 | STZ-induced diabetic mice | iv (tail vein) injection of naked pDNA | Reduced renal hypertrophy & albuminuria; Inhibited expression of TGF-β1 and MCP-1; Activation of STAT-1 & -3 |
Liu et al. (2012) | Megsin | STZ-induced diabetic CD-1 mice | iv (tail vein) injection of naked pDNA encoding anti-Megsin siRNA | Decreased proteinuria & collagen IV buildup in glomeruli; Reduced renal cell proliferation by regulating MMP-2 and tissue inhibitor of MMP-2 |
Tang et al. (2012) | Mitofusin-2 (Mfn-2) | STZ-induced diabetic rats | Intra-arterial (renal artery) injection of adenovirus vector | Reduced proteinuria; Improved enlargement of glomeruli & accumulation of ECM; Reduced synthesis of collagen IV & thickening of the basement membrane; No prevention of apoptosis; activation of p38 & ROS buildup were decreased |
Flaquer et al. (2012) | HGF | Female C57BLKS mice (db/db) | im injection of naked pDNA | Improved expression of SDF-1; Increased number of monocyte-derived macrophages contributing to renal tissue repair & regeneration |
Wang et al. (2014) | Histone deacetylase-4 (HDAC-4) | STZ-induced diabetic rats & db/db mice | Intraparenchymal injection of naked pDNA encoding anti-HDAC-4 siRNA | Prevented podocyte injury; Reduced renal dysfunction in diabetic rats through regulation of HDAC-1/STAT-1 signaling |
Yuan et al. (2014) | Globular adiponectin (ADPN) | STZ-induced diabetic rats | ip injection of naked pDNA | Decreased urine albumin excretion; Increased glomerular mesangial expansion; Decreased production of ROS; Prevented interstitial fibrosis; Promoted renal expression of eNOS & inhibited TGF-β1 expression |
Zhou et al. (2014) | Suppressor of cytokine signaling −2 (SOCS-2) | STZ-induced diabetic rats | iv (renal vein) injection of adenovirus vector | Reduced glomerular hypertrophy, hyper-filtration, inflammation, fibrosis & renal lesions; Decreased level of pro-inflammatory proteins such as TNF-α, IL-6 and MCP-1, and profibrotic proteins such as TGF-β, collagen IV & fibronectin |
Yang et al. (2014) | Thrombomodulin domain-1 (THBDD-1) | Male C57BLKs/J db/db mice | iv injection of adeno-associated virus vector | Improved albuminuria & renal function as well as renal interstitial inflammation & glomerular sclerosis by inhibition of mitochondria-derived apoptosis; Prevented activation of NF-κB pathway & NLRP-3 inflammasome & facilitated nuclear translocation of NRF-2 |
Fatal accidental deaths have been reported form studies after systemic administration of adenoviral vectors due to over activation of the innate immune response (Marshall 1999). Adeno-associated virus (AAV) has moved to the forefront as the most attractive viral vector. The recombinant form remains almost completely episomal, with only minimal genomic integration in mice (Inagaki et al. 2008) and humans (Kaeppel et al. 2013). While clinical trial experience is growing rapidly with recombinant AAV vectors, immune responses to the capsid and toxicity remain potential concerns. As a result, research into the development of non-viral vectors continues to evolve.
17.5.2 Non-viral Gene Delivery
The strong inflammatory response elicited by viral vectors during in-vivo gene delivery has boosted research into developing non-viral gene vectors that may be safer for clinical applications. New classes of chemical vectors and physical delivery systems have been developed which are economical; provide stable transfection with minimum immunogenicity. Although the transfection efficiency of non-viral vectors is lower compared to the viral vectors, they are believed to be attractive alternatives for their lack of specific immune response, versatility, ease of large-scale production and simplicity of usage. Non-viral based gene therapy can be broadly classified into chemical and physical methods.
17.5.2.1 Chemical Methods
Chemical methods utilize cationic lipids, cationic polymers and cell-penetrating peptides that can be synthesized to target specific cells locally or systemically. Chemical vectors overcome the safety concerns associated with disease-causing viral vectors and can also be custom engineered according to the purpose. The primary mechanism for non-viral transfection using chemical vectors is through endocytosis by the target cell (Hoekstra et al. 2007). Cationic lipids/liposomes have been designed to facilitate efficient binding of nucleic acid vectors to their surface. The efficiency of in-vivo transfection with cationic lipids/liposomes is highly dependent on the lipid structure, DNA properties and size (Alatorre-Meda et al. 2010; Liang et al. 2013). Although the association of nucleic acids to the cationic lipid is through electrostatic interaction, due to the lack of mechanisms for recognition of cell surface markers, in-vivo transfection with cationic lipids/liposomes is non-specific with remote off-target effects. Studies have implemented antibody-conjugated liposomes to improve cell specificity (Asgeirsdottir et al. 2008), but these are still unable to achieve organ specificity. Additionally, primary cells, progenitor cells and stem cells have proven more difficult to transfect using chemical vectors. Many viral and chemical vectors have limited efficiency in transfecting non-dividing cells. Thus, the limitations of non-viral cationic lipid vectors for in-vivo and in-vitro studies have encouraged development of newer physical method-based delivery techniques for increasing target specific transfection.
17.5.2.2 Physical Methods
The most common methods of physical gene transfer include electroporation and sonoporation. Electroporation can be defined as the application of an external electrical field to a cell population or a tissue for a fixed duration to increase cell permeability to nucleic acids and small proteins by forming localized transient pores in the cell membrane (Andre and Mir 2010). Electroporation has successfully transfected both dividing and non-dividing cells, which has been a major limitation of viral and chemical-based methods. Although electroporation may be able to efficiently transport nucleic acids into cells, this advantage usually comes at a cost of low cell viability. Low cell viability has been associated with irreversible damage to the cell membrane and increased cytotoxicity occurring due to changes in pH. Thus, an ideal gene delivery technique would require provision of: (1) Maximized transfection efficiency of the therapeutic gene, (2) good safety profile, (3) minimal invasion during gene delivery, (4) enhanced target specificity for improved localization of exogenous genes without off-target effects, (5) high cell viability, and (6) repeatability as required. Table 17.2 lists pre-clinical studies using non-viral vectors to combat DN.
Comparable to electroporation, high-power ultrasound has exhibited the ability to induce pore formation in cell membranes through cavitation mechanisms. This phenomenon has been termed sonoporation, and allows for transport of nucleic acids/proteins/drugs into the cell cytosol. Cavitation and subsequent tissue transfection has been shown to be enhanced with the use of ultrasound contrast agents, known as microbubbles. This combined delivery of therapeutic ultrasound and microbubbles, conjugated with gene vectors/antibodies/drugs, comes under several headings, including ultrasound-mediated gene delivery (UMGD, or ultrasound-targeted microbubble destruction (UTMD). As discussed earlier, this is a non-invasive and target-specific method of gene transfer that fulfills most of the requirements of an ideal gene therapy platform.
17.6 Ultrasound-Mediated Gene Delivery
UMGD is a unique gene delivery platform that results in targeted transfection with increased in-vivo transfection efficiency of non-viral gene vectors. This technique can achieve high organ-specific transfection, with minimum invasiveness and immunogenicity. Also referred to as ultrasound-targeted microbubble destruction, UMGD employs high power ultrasound and gene-bearing carrier microbubbles for achieving targeted gene delivery.
17.6.1 Microbubbles
The carrier agents for UMGD are microbubbles; very small, gas-filled bubbles. The gas-filled core is usually made up of a high molecular weight gas, such as decafluorobutane, perfluoropropane or sulfur hexafluoride, while the outer shell is composed of biocompatible lipids or proteins (Klibanov 2006). The low diffusivity and solubility properties of the heavy gas core allows for prolonged stability in circulation. The rheology of microbubbles is comparable to that of red blood cells, with a mean diameter of approximately 2–4 μm; giving them the ability to freely traverse through the microvasculature without being impeded (Lindner et al. 2002). Microbubble contrast agents have been conventionally used as a diagnostic contrast agent for clinical use with ultrasound (Mulvagh et al. 2008; Honos et al. 2007). The ability to modify microbubbles to become a carrier vessel for nucleic acid vectors, such as plasmid DNA, microRNA (miRNA) and silencing-RNA (siRNA) allows them to be used for UMGD.
17.6.2 UMGD: Methodology and Mechanisms
A schematic diagram of UMGD is depicted in Fig. 17.1. Firstly, microbubble-nucleic acid complexes are generated. This is either by direct incorporation of nucleic acids during microbubble sonication, by conjugation of nucleic acids (plasmid DNA, siRNAs, miRNAs or viral DNA) to the outer surface of microbubbles via electrostatic interactions (charge-coupling), or by simple mixing/co-administration. Secondly, the microbubble-nucleic acid complexes are administered intravenously. Thirdly, the complex circulates through the systemic vascular system during transmission of high power triggered ultrasound over the target tissue/organ. The destructive power of ultrasound causes acoustic disruption of the gas-filled microbubbles and facilitates the nucleic acid uptake by the endothelium and surrounding cells/interstitium. This uptake is via several mechanisms, including formation of microjets, induction of transient pores at the cellular membrane and endocytosis (Christiansen et al. 2003; Kodama et al. 2006; Meijering et al. 2009).

Fig. 17.1
Schematic diagram of ultrasound-mediated gene delivery (UMGD) to the kidney. Firstly, co-injection (intra-venous or intra-arterial) of nucleic acids (DNA) and microbubbles; Next, application of external high power ultrasound over the kidney(s); Bioeffects of ultrasound destruction within target tissue/organ leading to vascular and extravascular transfection, protein synthesis/secretion or knockdown, and resultant biological effect on diabetic nephropathy
While transfection efficiency of UMGD is relatively modest compared to viral vectors, studies using marker genes, such as luciferase, have demonstrated that transfection can be optimized by (1) using longer pulsing intervals for triggered delivery. This allows replenishment of the microvasculature by microbubble-DNA complexes within the target tissue/organ between destructive delivery pulses of ultrasound (Chen et al. 2003; Song et al. 2002). Conversely, continuous ultrasound transmission results in minimal transfection due to continual destruction of microbubble-DNA complexes as soon as they enter the ultrasound beam, (2) using higher acoustic powers for ultrasound application, with low transmission frequency and a greater mechanical index (Chen et al. 2003). This method must however be balanced against higher adverse bioeffects, especially when applied to the kidney (Williams et al. 2007), (3) the use of intra-arterial injections of carrier microbubbles versus intravenous delivery (Song et al. 2002; Christiansen et al. 2003) also results in greater tissue damage and vessel hemorrhage, and (4) use of repeated deliveries resulting in a more sustained duration of transgene expression (Bekeredjian et al. 2003).
17.6.3 UMGD: In-Vivo Applications
There have been numerous studies of UMGD in multiple organs, delivering gene therapy in many pre-clinical models of human disease. Studies have demonstrated the efficacy of UMGD for therapeutic angiogenesis in a rat model of peripheral arterial disease (PAD). UMGD of a plasmid DNA encoding VEGF and green fluorescent protein (GFP) in a bicistronic vector resulted in increased microvascular perfusion and vessel density (Leong-Poi et al. 2007). Using the same vector and PAD model, UMGD was more effective for therapeutic angiogenesis compared to direct intramuscular (im) injection of the VEGF/GFP plasmid, despite lower transgene expression. The higher efficiency of pro-angiogenic gene therapy by UMGD was likely due to its targeted vascular transfection, as compared to IM injections. While IM injections resulted in local transfection of myocytes and perivascular regions, UMGD resulted in more diffuse transfection of arteriole and capillary endothelium and surrounding myocytes (Kobulnik et al. 2009). UMGD can be effective for targeted cardiac transfection, including delivery of pro-angiogenic genes in pre-clinical models of myocardial infarction (Fujii et al. 2009, 2011) and heart failure (Lee et al. 2013). A major advantage of UMGD is its noninvasive nature, which allows for delivery of multiple genes through repeated gene therapy (Fujii et al. 2011; Smith et al. 2012). Using multiple UMGD deliveries of plasmid DNA encoding for stem cell factor and stromal cell derived factor-1 (SDF-1) in a rat model of myocardial infarction, Fujii et al. showed increased vascular density in the peri-infarct region and greater myocardial perfusion and ventricular function compared with untreated animals (Fujii et al. 2011). The use of UMGD for in-vivo gene transfer has been applied for targeted gene transfection in other organs and tissues that can be imaged by diagnostic ultrasound, including kidney, pancreas (Chen et al. 2006, 2007, 2010) and tumors (Fujii et al. 2013; Carson et al. 2011, 2012).
17.7 Ultrasound-Mediated Gene Delivery: Applications in Diabetic Nephropathy
Gene therapy offers a promising approach for the treatment of chronic kidney diseases; both inherited genetic disorders, like hereditary nephritis (Alport syndrome), and acquired diseases, including nephropathy. Despite promising results of numerous pre-clinical studies of gene therapy in animal models of kidney diseases, translation to the clinic has been remarkably slow. Major questions that limit clinical translation include choice of target gene, which vector, optimal route of delivery, what dose, how many doses and at what intervals between doses. A comprehensive discussion of the different routes, vehicles and delivery techniques for gene therapy has been carried out in Sect. 17.5.
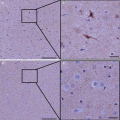
Studies have demonstrated transfection of the kidney using UMGD. Azuma and colleagues published the very first report of UMGD to the kidney in 2003 (Azuma et al. 2003). They used UMGD with Optison™ (a commercially available microbubble contrast agent, GE) to transfect fluorescein isothiocyanate (FITC) labeled NFκB-decoy into donor kidneys to prevent acute kidney rejection and prolong survival in a rat renal allograft model. They also used UMGD to transfect luciferase reporter gene to the kidney, showing that transfection occurred in 70–80 % of glomeruli and most tubular cells (Azuma et al. 2003). From a mechanistic standpoint, Zhang et al. (2014) demonstrated that the interactions of ultrasound (frequency 7 MHz, mechanical index = 0.9, peak negative acoustic pressure = 2.38 MPa) and microbubbles used for UMGD increases renal interstitial capillary permeability, as measured by Evan’s blue extravasation in rats with early DN induced with streptozotocin (STZ). Importantly, hemorrhage or necrosis was not observed. Figure 17.2 shows fluorescent confocal microscopic images of a control non-transfected kidney and a kidney where UMGD was performed. Initially, 2.5 μg of red fluorescent (Alexa Fluor BlockIT Red, Invitrogen) control microRNA was charge-coupled onto 1 × 109 cationic lipid microbubbles. Then this solution was intravenously administered during external application of intermittent high-powered ultrasound (frequency 5 MHz, 2 cm depth, power 120 V) at a pulsing interval of 10s (internal timer). UMGD resulted in fairly diffuse transfection throughout the kidney parenchyma, including glomeruli and tubules.

Fig. 17.2
In-vivo transfection by UMGD to the kidney. UMGD of fluorescent-labeled control miRNA (red) to the kidney via a phased array transducer. A transmission frequency of 5 MHz at 120 V and pulsing interval of 5 s were used. In-vivo UMGD delivery of fluorescent-labeled oligonucleotides to the kidney resulted in high and diffuse in-vivo transfection (Top panels), as compared to control non-delivered kidneys (Bottom panels)
Preclinical studies using UMGD have shown benefits in various models of kidney disease, including DN, using multiple different gene targets. Table 17.3 provides important details on all studies to date of UMGD in a variety of pre-clinical models of kidney disease, many of which targeted DN. Lan et al. (2003) investigated UMGD to specifically target the transforming growth factor (TGF)-β/Smad pathway, and subsequently renal fibrosis in a rat unilateral ureteral obstruction model. Ultrasound-mediated doxycycline-regulated Smad7 gene transfer resulted in increased Smad7 expression and inhibited tubulointerstitial fibrosis, thus providing a novel therapeutic strategy against renal fibrosis. For UMGD, Optison™ microbubbles in saline were mixed with 25 μg of chosen plasmid, and then 0.5 mL of the mixture was injected into the left renal artery. The ultrasound transducer (Ultax UX-301; Celcom Medico Inc., Japan) was placed directly onto one side of the left kidney, with a continuous-wave output of 1-MHz ultrasound at 5 % power output, for a total of 60 s with 30 s intervals.
Table 17.3
Preclinical UMGD studies in kidney disease
Reference | Gene/target | Animal model | Ultrasound settings | Microbubble | Major findings |
|---|---|---|---|---|---|
Azuma et al. (2003) | NF-κB | Wistar-Lewis rat renal allograft model | 2 MHz 2.5 W/cm2 30 s – 8 min | Optison | Inhibited action of inflammatory molecules associated with graft rejection; preserved histological structure; indication of prolonged graft survival & prolonged animal survival compared to controls |
Lan et al. (2003) | Smad-7 | Rat unilateral ureteral obstruction model | 1 MHz 5 % power out 60 s with 30 s intervals | Optison | Inhibited tubulointerstitial fibrosis; Resulted in complete inhibition of Smad-2 & -3 action, thus provided a novel therapeutic strategy against renal fibrosis |
Hou et al. (2005) | Smad-7 | Rat remnant kidney model | 1 MHz 5 % power output 60 s with 30 s intervals | Albumin perflutren-filled microbubbles | Reduced renal & vascular sclerosis; Inhibited Smad-2 & -3 activation; Prevented progressive renal injury by inhibiting the rise of proteinuria & serum creatinine
Related posts: Heat-Based Tumor Ablation: Role of the Immune Response Heat-Based Tumor Ablation: Role of the Immune Response
 Design of Microbubbles for Gene/Drug Delivery Design of Microbubbles for Gene/Drug Delivery
 Microbubble-Assisted Ultrasound for Drug Delivery in the Brain and Central Nervous System Microbubble-Assisted Ultrasound for Drug Delivery in the Brain and Central Nervous System
 Co-administration of Microbubbles and Drugs in Ultrasound-Assisted Drug Delivery: Comparison with Drug-Carrying Particles Co-administration of Microbubbles and Drugs in Ultrasound-Assisted Drug Delivery: Comparison with Drug-Carrying Particles
 Stimulation of Bone Repair with Ultrasound Stimulation of Bone Repair with Ultrasound
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|