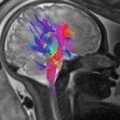
Fig. 7.1
Macrocephaly. (a, b) Sagittal and coronal T2 single-shot haste (SSH) MR images demonstrate an enlarged head secondary to a large left-sided extra-axial hematoma (white arrows)
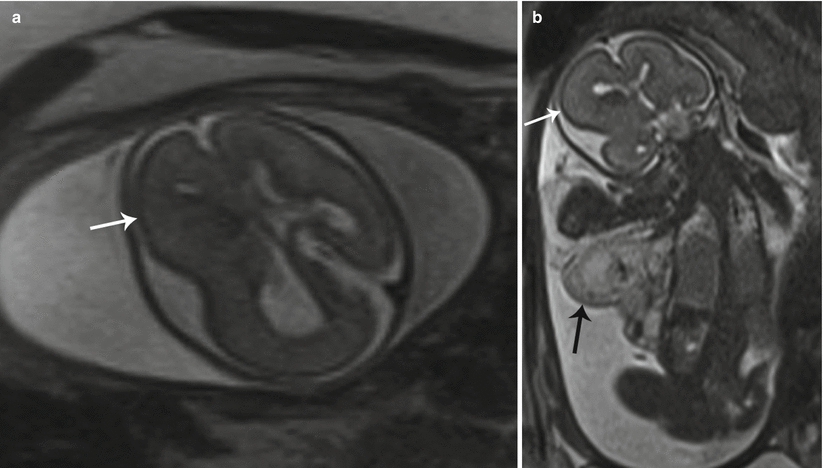
Fig. 7.2
CLOVES syndrome. (a, b) Axial and coronal T2 SSH MR images of the fetal head demonstrate hemihypertrophy of the right cerebral hemisphere (white arrows). A large venolymphatic malformation (black arrow) of the right axilla was also noted in this patient with CLOVES syndrome
7.2.2 Microcephaly
Microcephaly is a manifestation of an underlying small brain which may be secondary to arrest of growth or due to volume loss from an insult such as infection, ischemia, or hemorrhage (Fig. 7.3a, b). Causes of growth arrest are numerous relating to drugs, maternal disease, and genetic/syndromic disorders among other causes.
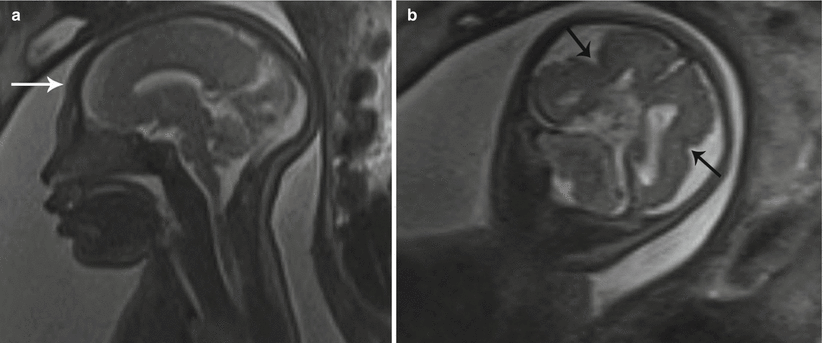
Fig. 7.3
Microcephaly. (a) Sagittal T2 SSH MR image of the fetal brain demonstrates a small cranium (white arrow). (b) Coronal T2 SSH MR of this patient’s brain revealed bilateral perisylvian polymicrogyria (black arrows)
7.2.3 Abnormal Skull Shape
Abnormal calvarial configuration has a number of causes, many of which are related to genetic abnormalities and may have fairly characteristic appearances [3]. A “lemon” shape of the skull has been identified in association with neural tube defects due to loss of subarachnoid spaces with collapse of calvarial bones around the brain (Fig. 7.4). A more triangular “strawberry” configuration is described in association with trisomy 18. Following fetal demise, collapse of the brain may result in overlapping calvarial bones termed Spalding sign. Cases of severe oligohydramnios or anhydramnios may also result in abnormal calvarial morphology, with configuration of the distortion varying depending on the resultant distribution of external pressure.
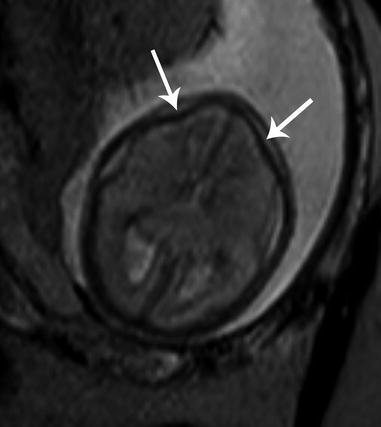
Fig. 7.4
Lemon-shaped head. Axial T2 SSH MR image illustrates flattening of the bilateral frontal bones (white arrows) in the setting of Chiari II malformation
Craniosynostoses are a specific collection of skull abnormality defined by premature fusion of 1 or more cranial sutures, which prevents calvarial growth in planes perpendicular to the abnormally fused suture(s). A characteristic morphologic abnormality of the skull and face corresponds to each specific fusion location. For example, the most common form of craniosynostosis involves premature fusion of the sagittal suture resulting in generalized elongation of the calvarium with narrowing of the skull width, termed scaphocephaly or dolichocephaly [5]. Coronal craniosynostosis may result in plagiocephaly or brachycephaly, if bilateral, while metopic craniosynostosis results in trigonocephaly (Fig. 7.5). Craniosynostoses may exist independently or as part of a syndrome. Syndromic craniosynostoses include Apert, Crouzon, Pfeiffer, Saethre–Chotzen, Jackson–Weiss, and Antley–Bixler. Apert syndrome is characterized by coronal craniosynostosis with possible involvement of other sutures, midface hypoplasia, and syndactyly of the hands and feet (mitten hands) [6, 7].
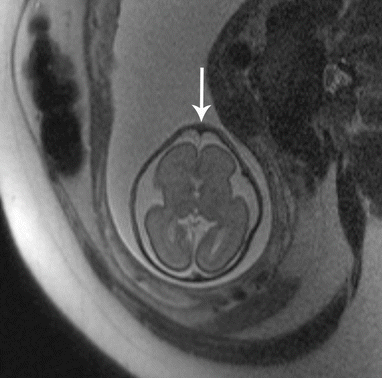
Fig. 7.5
Trigonocephaly. Axial T2 SSH MR image demonstrates a triangular appearance of the forehead secondary to underlying metopic synostosis (white arrow) (Courtesy of Dr. Tamara Feygin, CHOP)
7.3 Scalp Masses
There are numerous causes of fetal scalp masses, the most common being hemangiomas, lymphatic malformations, and congenital inclusion cysts such as dermoids/epidermoids. These masses are not limited strictly to the scalp and are discussed in detail below. Cephalocele is another potential cause of a fetal scalp mass and is an entity in which MRI can be of particular benefit as it may both confirm a calvarial defect (which sonography may falsely detect due to scanning angle) and also define the intracranial anatomy. Evaluation of intracranial anatomy is especially helpful in confirmation of diagnosis of a cephalocele with identification of associated venous sinus anomalies (Fig. 7.6a, b) [8].
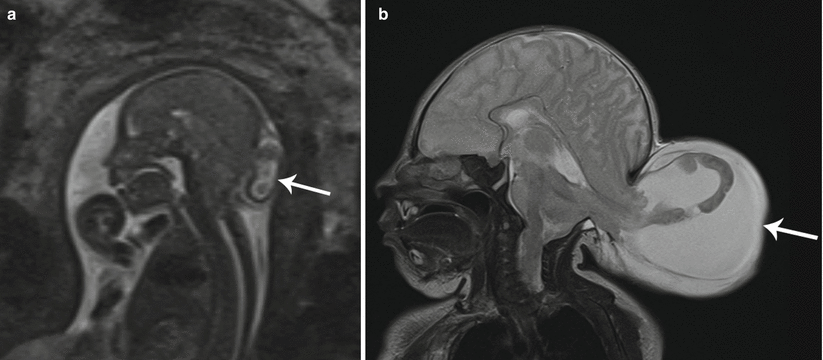
Fig. 7.6
Occipital encephalocele. (a) Sagittal T2 SSH MR image shows a midline occipital calvarial defect with herniation of brain and meninges (white arrow). (b) Postnatal sagittal T2-weighted MR image confirms the prenatal findings (white arrow)
7.4 Abnormal Orbits/Globes
7.4.1 Hypotelorism
Hypotelorism is characterized by eyes which are abnormally close together. This can be quantified by measurement of interocular diameter (IOD: medial-to-medial margin of the vitreous) and binocular diameter (BOD: lateral-to-lateral margin of the vitreous) below the 5th percentile for gestational age (Fig. 7.7a, b) [9]. Caution should be used in comparing only to standardized growth charts specifically for MRI measurements, as ultrasound measurements rely upon the bony margins rather than vitreous margins, resulting in larger BOD and smaller IOD when calculated with ultrasound compared to MRI [9, 10]. An alternative simplified approach to recognizing hypotelorism is to compare the IOD to a single orbital width, both of which should be roughly equal.
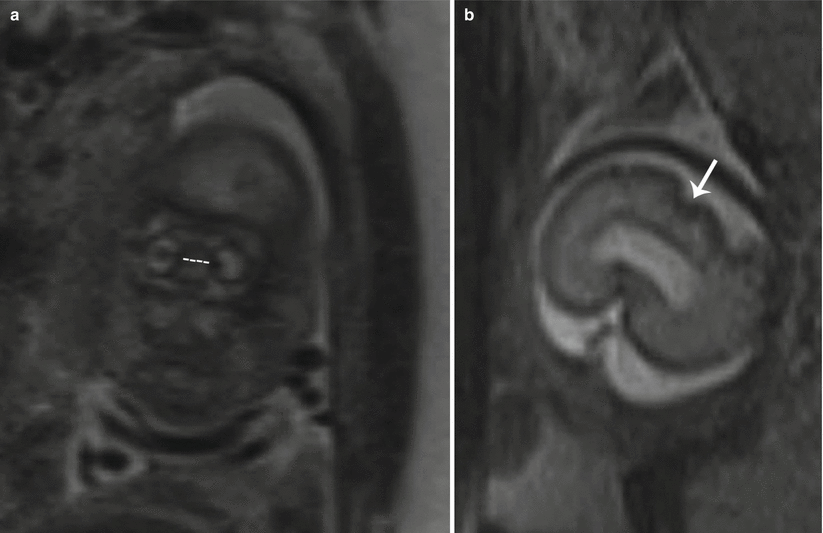
Fig. 7.7
Hypotelorism. (a) Coronal T2 SSH MR image demonstrates a shortened interocular distance (dotted line) in a fetus with trisomy 13. (b) Axial T2 SSH MR image of this patient’s brain revealed lack of normal forebrain cleavage and a monoventricle indicative of holoprosencephaly (white arrow)
As over half of these patients have an underlying aneuploidy, hypotelorism identified on fetal MRI should prompt evaluation for intracranial anomalies. Often associated are midline malformations of the brain such as holoprosencephaly. Secondary hypotelorism may be caused by abnormal calvarial development, such as metopic synostosis or microcephaly [11].
7.4.2 Hypertelorism
Hypertelorism, contrastingly, is characterized by eyes which are abnormally wide set. This is quantified by IOD and BOD above the 95th percentile for gestational age [9]. Similarly, hypertelorism can be inferred when the IOD is larger than the orbital width.
Primary hypertelorism is rarely present in isolation and often is accompanied by an underlying chromosomal derangement or syndrome. Secondary hypertelorism may result due to craniosynostoses, anterior encephalocele (Fig. 7.8a, b), or midline facial masses [11]. Maternal intake of antiepileptic drugs has also been suggested as a cause of hypertelorism.
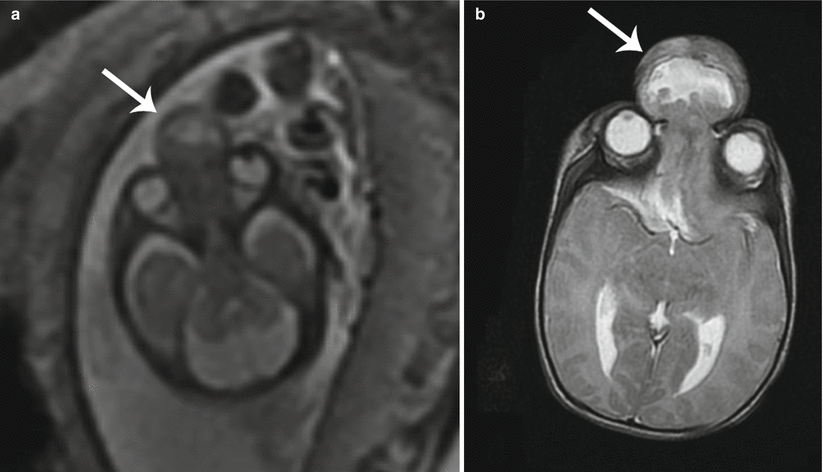
Fig. 7.8
Hypertelorism. (a) Axial T2 SSH MR image shows a frontonasal encephalocele (white arrow) with consequent widening of the interocular distance. (b) Postnatal axial T2-weighted MR image confirms the findings (white arrow)
7.4.3 Proptosis
Proptosis is defined by anterior displacement of the globes. This appearance may be caused by shallow orbits in association with a craniosynostosis. Rarely, proptosis may be the presenting feature of an underlying orbital mass or orbital encephalocele [3].
7.4.4 Microphthalmia/Anophthalmia
Microphthalmia is characterized by a small globe below the fifth percentile in size for gestational age. Severe microphthalmia may be mistaken for anophthalmia, a far less common entity with complete absence of the globe and the neuroectodermal tissue of the orbit. True anophthalmia may occur from failure of formation of the optic vesicle and requires preservation of non-neuroectodermally derived tissue, such as the eyelids, conjunctiva, lacrimal apparatus, and extraocular muscles; a small cystic structure may be present within the orbit that does not represent a small globe. Fetal MRI is useful in determining the degree of microphthalmia and delineating the internal structure of the globe and orbital contents in addition to assessing for associated brain findings (Fig. 7.9) [12, 13].
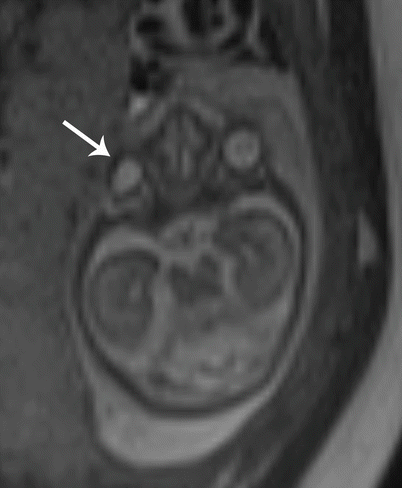
Fig. 7.9
Microphthalmia. Axial T2 SSH MR image demonstrates a small right eye (white arrow) and associated hypertelorism in this patient with the genetic diagnosis of Fasier syndrome
Microphthalmia/anophthalmia is a feature of many hereditary conditions and is frequently seen with aneuploidy, such as trisomy 13 (Patau syndrome). They are also a feature of various syndromes such as Aicardi syndrome, CHARGE syndrome (coloboma, heart defects, choanal atresia, retarded growth and development, genital anomalies, and ear anomalies), Walker–Warburg syndrome, and oculocerebrocutaneous syndrome. Often the syndrome is unknown and a diagnosis of multiple congenital anomalies (MCA) is applied. Microphthalmia/anophthalmia may also be acquired with etiologies including congenital infection, fetal alcohol syndrome, and congenital cataracts.
7.4.5 Optic Disc Coloboma
Optic disc coloboma is characterized by a focal defect of the posterior globe at the site of optic nerve head insertion resulting in outpouching of the vitreous fluid. Coloboma may be sporadic or inherited, with the former typically unilateral and the latter typically bilateral. Coloboma may be seen in association with numerous syndromes including CHARGE syndrome, Walker–Warburg syndrome, Aicardi syndrome, Goldenhar syndrome, and Noonan syndrome [14]. Imaging findings consist of a focal outpouching of high-signal vitreous on T2-weighted imaging at the optic nerve insertion site (Fig. 7.10a, b). A retrobulbar “cyst” that communicates with globe may be present. Optic disc coloboma may be associated with microphthalmia and, at times, retinal detachment [15].
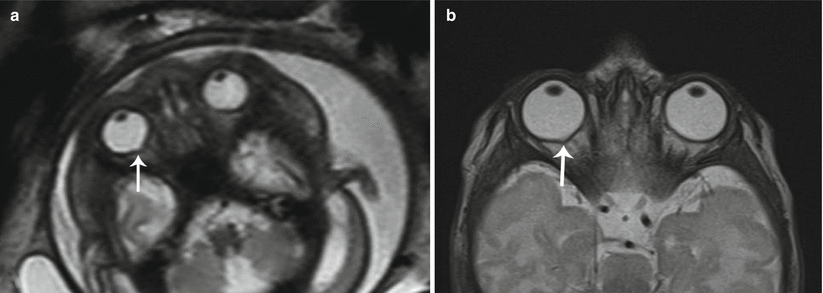
Fig. 7.10
Coloboma. (a) Axial T2 SSH MR image suggested a small outpouching along the posterior aspect of the right eye (white arrow) in this fetus that was otherwise normal. (b) Postnatal axial T2-weighted MR image confirms the suspicion for coloboma (white arrow)
7.4.6 Persistent Hyperplastic Primary Vitreous
Persistent hyperplastic primary vitreous (PHPV), or persistent fetal vasculature, results from a failure of gestational involution of the embryonic hyaloid vascular system within Cloquet’s canal. A triangular retrolental soft tissue mass remains, which, in the majority of cases, extends posteriorly to the optic disc. It is usually isolated and unilateral. Bilateral lesions may be associated with trisomy 13 or syndromes such as Norrie and Walker–Warburg [16]. Postnatally, children with PHPV present with leukocoria and may go on to develop glaucoma, cataract, intraocular hemorrhage, or retinal detachment. As PHPV may be often confused with retinoblastoma, CT imaging may be employed to detect intralesional calcifications which are rare in PHPV.
7.4.7 Neoplastic and Nonneoplastic Masses of the Orbits
Primary tumors of the fetal orbit are rare, with reports of retinoblastoma, teratoma, and sarcoma. More commonly, the orbit is secondarily affected by extension of an intracranial or cervical neoplasm. Depending upon the size and directional growth of the tumor, the globe may be completely obscured and the orbit may be destroyed. MRI is helpful in determining intracranial extension.
While retinoblastoma represents the most common intraocular malignancy of childhood, prenatal detection of congenital retinoblastoma is limited. The orbit is a rare location for primary teratoma development. Orbital teratomas are characteristically massive tumors which appear as complex cystic and solid masses on imaging. Despite being benign tumors, teratomas are locally aggressive and result in severe facial deformity. General characteristics of teratomas are discussed in further detail below under “Cervical Teratomas” and “Epignathus.”
At the most benign end of the spectrum, a dacryocystocele, or lacrimal duct cyst, may in rare instances resemble a cystic orbital mass. In the axial plane, it appears as a round cyst resembling an “extra eye.” In the coronal and sagittal planes, it can be tubular in configuration when there is nasal extension. Though typically unilateral, dacryocystoceles may be bilateral. Bilateral involvement with nasal extension has been reported to cause nasal obstruction soon after birth. Pathologically, it results from nasolacrimal duct obstruction with distention of the duct and sac. As an isolated finding, these are almost always sporadic. Most resolve without surgery, either in utero or within the 1st year of life.
7.5 Cleft Lip and Palate
Cleft lip is the most common fetal facial anomaly, occurring in 1 in 700 live births. While the two may occur separately, cleft lip is accompanied by cleft palate in 50–85 % of affected patients [4, 17]. Orofacial clefting primarily results as a consequence of failed fusion of the facial mesenchyme during embryogenesis. When complete, the cleft extends through the upper lip to involve the hard palate. Clefts may be unilateral (left more common than right), bilateral, or midline, with respectively increasing rates of associated aneuploidy (Fig. 7.11a, b). Complex facial clefts, not conforming to the pattern of developmental clefts, may occur with amniotic band syndrome (Fig. 7.12a, b).
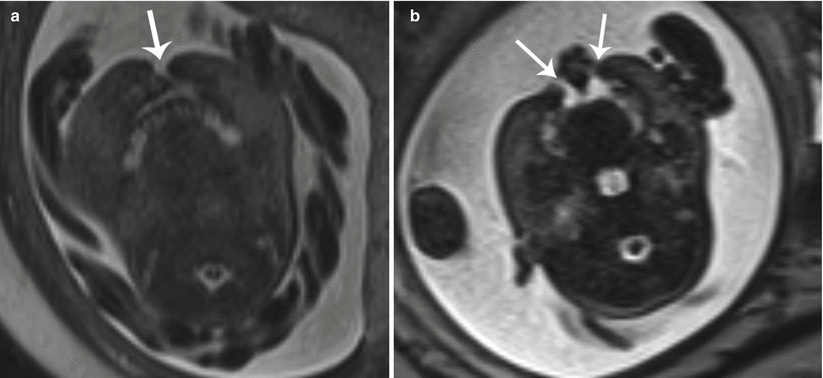
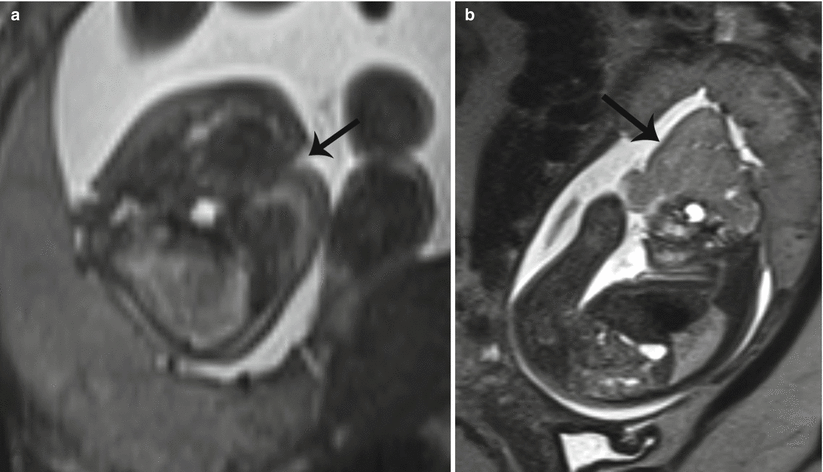
Fig. 7.11
Cleft lip/palate. (a) Axial T2 SSH MR image demonstrates a unilateral cleft lip (white arrow). (b) In a different fetus, bilateral cleft lip/palate (white arrows) with an intervening triangular segment was observed on this axial T2 SSH MR image
Fig. 7.12
Complex cleft lip/palate. (a) Axial T2 SSH MR image shows an atypical slash defect of the lip and palate (black arrow) in a patient with amniotic band syndrome. (b) This patient also exhibited exencephaly with presence of dysmorphic brain material (black arrow)
Fetal MRI is very good at defining an amniotic fluid-filled cleft [18]. The multiplanar capability of MRI also makes it particularly useful in visualizing the posterior soft palate and in determining overall extent of involvement for surgical planning [19, 20]. However, unilateral cleft lip without cleft palate, as well as closely apposed cleft orofacial cleft, can nonetheless be difficult to visualize on fetal MRI. Bilateral orofacial cleft has a characteristic premaxillary protrusion on profile view resulting from elevation of the median nasal prominence; in the axial plane, a midline triangular tissue lies between the two clefts.
More than 400 syndromes are associated with orofacial clefting; the most common associated chromosomal abnormality is trisomy 13 and trisomy 18 [4, 17]. Various infections and teratogens have also been implicated as the underlying etiology of this deformity. Midline orofacial clefts are often associated with midface hypoplasia. Additionally, midline defects have an increased association with holoprosencephaly and as such should prompt careful scrutiny of the intracranial contents [3].
7.6 Abnormal Mandible
7.6.1 Micrognathia
Micrognathia, or mandibular hypoplasia, is related to a defect in the first and second branchial arches. It is typically coupled with retrognathia, abnormal posterior positioning of the mandible with receding chin. This deformity can be subjectively observed but is further enhanced with calculation of an inferior facial angle <50°, by calculating the intersection of a line orthogonal to the forehead and another drawn from the tip of the mentum to the anterior border of the upper lip [21]. It may develop in isolation or most often is present in conjunction with a variety of conditions including Goldenhar syndrome (hemifacial microsomia), Pierre Robin sequence, and Treacher Collins syndrome [22]. There is a high association with chromosomal abnormalities involving up to 66 % of fetuses with micrognathia, commonly trisomy 13 and trisomy 18. Given the common association with other abnormalities, identification of micrognathia on fetal MRI may raise concern to closely evaluate for an underlying syndrome or genetic abnormality, which can be of importance in genetic counseling for the parents (Fig. 7.13a, b).
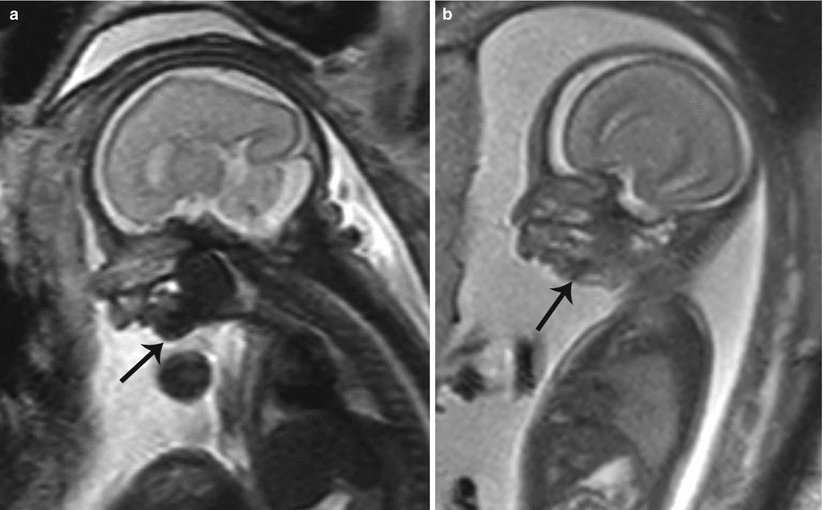
Fig. 7.13
Micrognathia. Sagittal T2 SSH MR images demonstrate (a) mild and (b) severe mandibular hypoplasia (black arrows) in two different fetuses
Caution should be taken to avoid overdiagnosis in early imaging; the mandible may often normally appear small in younger gestational age fetuses, as significant mandibular growth occurs during the third trimester. Additionally, asymmetric angulation of acquisition can give a false appearance of a small mandible [3].
Micrognathia yields a decreased size of the oral cavity with resultant posterior and superior displacement of the tongue and incomplete palatal fusion. Affected children frequently have impaired swallowing; as such polyhydramnios can be the initial presentation. As they are at risk for airway compromise, many of these patients require scheduled deliveries using the ex utero intrapartum treatment (EXIT) procedure.
7.6.2 Agnathia
Agnathia is an exceedingly rare and typically fatal malformation which is also related to a defect in the first branchial cleft. It is commonly associated with microstomia (small mouth), absent tongue, and midfacial location of the ears (Fig. 7.14a, b). A spectrum of concurrent abnormalities can occur including holoprosencephaly, genitourinary anomalies, cardiovascular anomalies, skeletal malformations, and situs inversus [23].
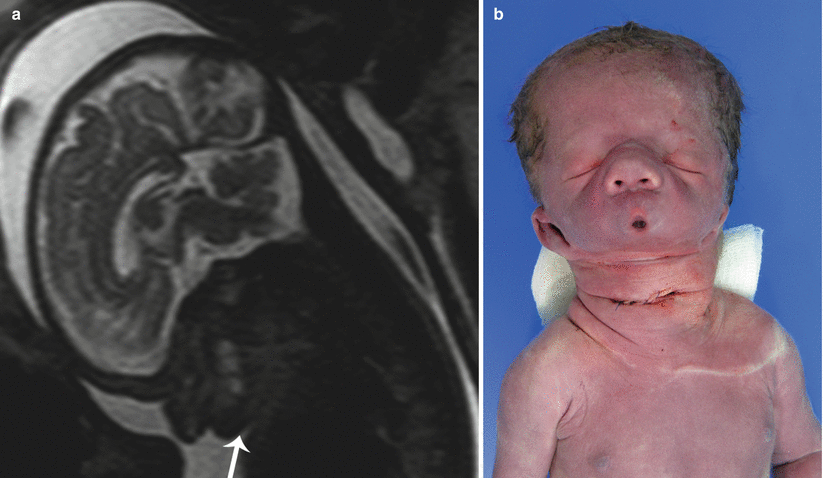
Fig. 7.14
Agnathia. (a) Sagittal T2 SSH MR image in a fetus with complete absence of the mandible (white arrow). Only the primordial teeth of the maxilla are seen. (b) Postmortem photograph of the same fetus demonstrates agnathia, microtia, a broadened nose, low-set abnormal ears, and slanted palpebral fissures
7.7 Lesions of the Oral Cavity
While masses of the fetal oral cavity are rare, they are of concern due to the high risk of airway obstruction and impairment of swallowing. This can result in polyhydramnios and/or respiratory distress at delivery [24]. As such, many of these patients may require delivery via the EXIT procedure with a pediatric head and neck surgeon present at time of delivery. Such lesions include congenital epithelial inclusion cysts (such as dermoid and epidermoid cysts), enteric duplication cysts, ranulas, teratomas (epignathus), epulis, and venolymphatic malformations, all of which are discussed in further detail below.
7.8 Congenital Lesions of the Neck
Congenital lesions of the neck detected in utero are overall very rare. These are varied in etiology and can be cystic (such as branchial anomalies, thymic cysts, meningocele, esophageal atresia, and thyroglossal duct cyst), solid (such as ectopic thymus, hemangioma, and sarcoma), or mixed cystic/solid (such as neuroblastoma and teratoma, both of which may also be completely solid) in composition.
7.8.1 Branchial Anomalies
Defects in the embryogenesis of the branchial apparatus may manifest as sinuses, fistulas, or cysts, the latter of which may be seen in conjunction with the former two. Prenatal diagnosis of branchial apparatus anomalies is very rare with few cases described in the literature [25–28].
Branchial cleft cysts are hypointense on T1-weighted sequences and hyperintense on T2-weighted sequences. If initially detected on US, they may appear as round/ovoid anechoic or hypoechoic thin-walled cysts often with movement of internal echoes. They may be located in a number of different anatomic locations, depending upon the branchial cleft of origin, and may rarely lead to airway obstruction. The most common lesion is a second branchial cyst, in the anterolateral neck (more frequently on the left), anterior to the sternocleidomastoid, posterior to the submandibular gland, and lateral to the carotid sheath [25]. First branchial cysts are located in the preauricular region or about the angle of the mandible. Third and fourth branchial cysts are exceedingly rare. Branchial cleft cysts are usually isolated lesions; however, they have been reported with branchio-oto-renal syndrome [29].
7.8.2 Cervical Thymic Cyst
Thymic cysts are remnants of the thymopharyngeal tracts which may be encountered in the neck, thoracic inlet, or mediastinum [30]. The thymic buds migrate inferiorly to form the thymopharyngeal ducts, which extend along the carotid sheath from the angle of the mandible to the superior mediastinum with attachment to the pericardium. In addition to thymic cysts, rests of ectopic thymic tissue can be found along the normal path of descent [31].
Thymic cysts are very rare and infrequently diagnosed in utero (Fig. 7.15a, b). They are commonly multilocular, but may be unilocular with size ranging from 1.4 to 8 cm. Internal signal is typically hypointense on T1-weighted images and hyperintense on T2 but may be heterogeneous if complicated by hemorrhage [32]. They are more common on the left and are located along the carotid space, splaying the carotid artery and jugular vein [31]. Up to 50 % have a mediastinal connection. Differential diagnoses include lymphatic malformation, thyroid cyst, thyroglossal duct cyst, and branchial cleft cyst.
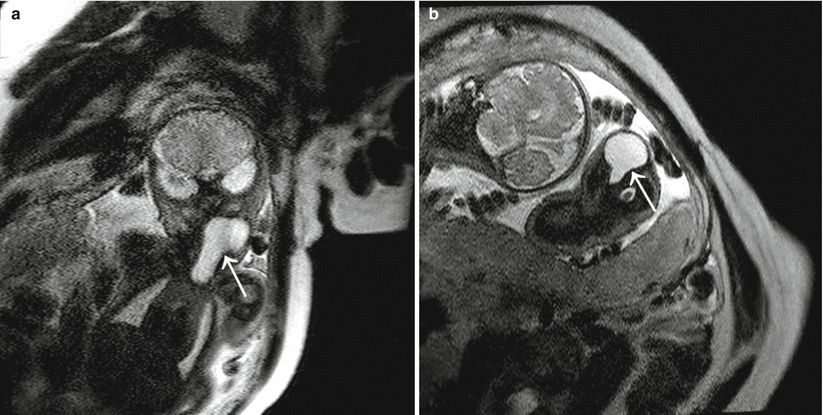
Fig. 7.15
Cervical thymic cyst. (a, b) Coronal and axial SSH MR images of a twin pregnancy reveal a bilobed cystic mass extending from the anterior mediastinum into the left neck along the course of the thymopharyngeal duct in twin A (white arrows) (Courtesy of Dr. Christopher Cassady, TCH)
7.8.3 Ranula
A ranula is a mucocele or retention cyst of a sublingual gland or a minor salivary gland duct located at the floor of the mouth which may extend into the submandibular/cervical region. They are classified according to location into either simple (intraoral) or plunging (oral/cervical) types. They have well-delineated borders and demonstrate internal low-T1- and high-T2-weighted signal. Congenital ranulas may elevate the floor of the mouth, displacing the tongue and rarely obstructing the airway [33, 34]. Differential diagnosis includes lymphatic malformation, thyroglossal duct cyst, and dermoid because of midline/paramidline positioning.
7.8.4 Thyroglossal Duct Cyst
While thyroglossal duct cysts represent the most common midline cervical anomaly, constituting 70 % of all congenital neck masses, prenatal diagnosis is rare [35, 36]. The exact incidence is unknown; however, 7 % of the population has been shown to have a thyroglossal duct remnant [36].
Prior to its involution, the embryologic thyroglossal duct extends from the foramen cecum of the tongue, along the anterior surface of the hyoid to the pyramidal lobe of the thyroid. Persistence of the duct results in cyst or sinus formation at any point of descent; infrequently, ectopic thyroid tissue can be found in the path of descent. The majority of duct remnants are midline or parasagittal, adjacent to the hyoid bone [37]. Rarely, these cysts are found within the tongue or the floor of the mouth, with the potential for airway obstruction [35]. The lesion can be occasionally associated with Cowden syndrome.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


