CHAPTER 43 Multiple Sclerosis and Other Idiopathic Inflammatory-Demyelinating Diseases
Idiopathic inflammatory-demyelinating diseases (IIDDs) represent a broad spectrum of central nervous system (CNS) disorders that can be differentiated on the basis of severity, clinical course, and lesion distribution, as well as imaging, laboratory, and pathologic findings. The spectrum includes monophasic, multiphasic, and progressive disorders ranging from highly localized forms to multifocal or diffuse variants.
Relapsing-remitting and secondary progressive multiple sclerosis (MS) are the two most common forms of IIDDs.1 MS can also have a progressive course from onset (primary progressive and progressive relapsing MS). Some patients have a benign course with minimal or no disability years after onset (benign MS). Fulminant forms of IIDDs include a variety of disorders that have in common the severity of the clinical symptoms, an acute clinical course, and atypical findings on MRI. The classic fulminant IIDD is Marburg disease. Balo’s concentric sclerosis, Schilder’s disease, and acute disseminated encephalomyelitis (ADEM) can also present as acute and severe attacks.
Monosymptomatic IIDD, such as transverse myelitis, optic neuritis, and brain stem demyelinating syndromes are commonly the first manifestation of MS, although a percentage of patients never actually develop the disease. Patients with monofocal clinical syndromes and MRI evidence of brain lesions consistent with demyelination have an 88% chance of developing clinically definite MS over the subsequent 14 years, whereas clinically similar patients with normal brain MRIs have less than a 20% chance of such progression.2 Hence, brain MRI is essential to target patients at high risk of early development of MS to select them for early immunomodulatory treatment.
MULTIPLE SCLEROSIS
Epidemiology
MS is one of the most common neurologic disorders and the second cause of disability in Western countries in young white adults. It is relatively common in Europe, the United States, Canada, New Zealand, and parts of Australia but rare in Asia and in the tropics and subtropics of all continents. Within regions having a temperate climate, the incidence and prevalence of MS increase with latitude, both north and south of the equator. Multiple sclerosis is twice as common in women as in men; men have a tendency for later disease onset with a poorer prognosis. The incidence of MS is low in childhood, increases rapidly after the age of 18, reaches a peak between 25 and 35, and then slowly declines, becoming rare at 50 and older.3 The disease affects approximately 1 million people between 17 and 65 years of age worldwide.
The environmental epidemiology of MS is poorly understood. Although a direct causal link with diverse infectious agents has been suggested, such infectious agents may simply provide the appropriate milieu for the development of an autoreactive immune response directed against CNS myelin. Up to now, only Epstein-Barr virus (EBV) infection has been established as a strong, consistent risk factor. As compared with uninfected individuals, MS risk is about 10-fold higher among individuals who experienced an undiagnosed EBV infection in childhood and at least 20-fold higher among individuals who developed mononucleosis.3
Clinical Presentation
Relapses or attacks typically present subacutely, with symptoms developing over hours to several days, persisting for several days or weeks, and then gradually abating. With time, the extent of recovery from these attacks often decreases, and baseline neurologic disability accumulates. The outcome in patients with relapsing forms of MS varies. About 50% of MS patients become dependent on a walking aid after 15 years of disease, whereas 10% remain free of any major disability after 25 years. The biologic basis of this variation in long-term clinical outcome is poorly understood. Neither symptom onset, nor gender, nor age at onset of symptoms predicts progression. However, increased attack frequency and poor recovery from attacks in the first year of clinical disease predict more rapid deterioration.4 The number of T2-weighted (T2W) lesions on brain MRI also predicts attack frequency and long-term disability.2
Relapsing-Remitting and Secondary Progressive Multiple Sclerosis
Over the following years, patients usually experience episodes of acute worsening of neurologic function, followed by variably complete recovery (relapsing-remitting course). Clinical and subclinical activity is frequent in this form. After several years of the relapsing-remitting course, more than 50% of untreated patients will develop progressive disability with or without occasional relapses, minor remissions, and plateaus (secondary progressive course).1
Primary Progressive and Progressive-Relapsing Multiple Sclerosis
Primary progressive forms comprise approximately 10% of MS cases. This form of MS begins as a progressive disease with occasional plateaus and relapses and temporary minor improvements. Progressive-relapsing MS follows a progressive course like primary progressive MS but shows clear acute relapses that may or may not be followed by full recovery.1
As compared with patients with the more common relapsing form of MS, patients with primary progressive MS tend to be older, show more rapid progression from onset, and are as likely to be male as female. The most common presentation by far is slowly progressing spastic paraparesis and, less frequently, progressive cerebellar, brain stem, visual, hemiplegic, and cognitive syndromes.5
According to the 2010 McDonald criteria (Table 43-1),6 a diagnosis of primary progressive MS can be established if patients show insidious neurologic progression for at least 1 year associated with MS-like abnormalities on brain and/or spinal MRI, and/or oligoclonal bands on cerebrospinal fluid (CSF) study. In patients who present with progressive spastic paraparesis, it is mandatory to exclude structural or metabolic causes of myelopathy by appropriate laboratory studies and spinal MRI.
TABLE 43-1 Diagnosis of MS with Disease Progression from Onset
CSF, cerebrospinal fluid.
From Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2010; 69:292–302.
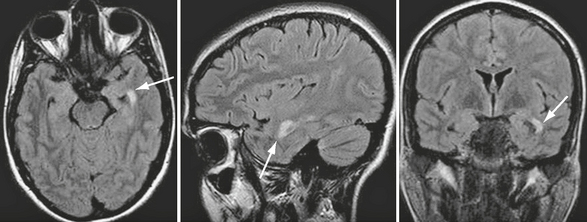
Compared with patients with the more frequent relapsing forms of MS, patients with primary progressive MS have smaller T2 lesion loads, smaller T2 lesions, slower rates of new lesion formation, and minimal gadolinium enhancement on brain MRI, despite their accumulating disability. The presence of extensive cortical damage, diffuse white matter tissue damage, and prevalent involvement of the spinal cord may partially explain this discrepancy between the MRI abnormalities and the severity of the clinical disease (Fig. 43-1).
FIGURE 43-1 Primary progressive MS. Transverse fast-FLAIR and FSE T2W sagittal MR images of the brain and cervical cord. The patient’s severe disability (paraparesis) cannot be explained by the few demyelinating brain lesions but can result from the multiple demyelinating lesions within the cervical cord.
Benign Multiple Sclerosis
Patients with benign MS have few new or enlarging lesions on serial brain MRI studies, and such lesions that occur have a lower incidence of enhancement after use of a contrast agent (Fig. 43-2), as compared with the typical relapsing-remitting forms of MS associated with progressive disability (Fig. 43-3). Prediction of a benign MS course may have an impact on the decision to initiate immunomodulatory medication, because this treatment may be unnecessary or might at least be postponed for many years.
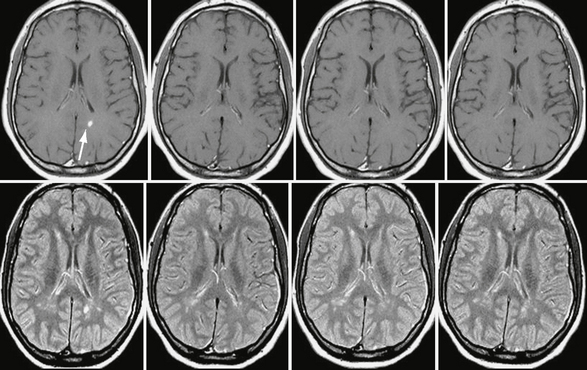
FIGURE 43-2 Benign MS. Serial, contrast-enhanced T1W (top row) and T2W (bottom row) MR images of the brain obtained yearly in a patient with benign MS. Note the small number of new lesions that developed over the 3-year follow-up and the very low incidence of contrast enhancement (arrow in the baseline scan).
(From Cañellas AR, Gols AR, Izquierdo JR, et al. Idiopathic inflammatory-demyelinating diseases of the central nervous system. Neuroradiology 2007; 49:393-469.)
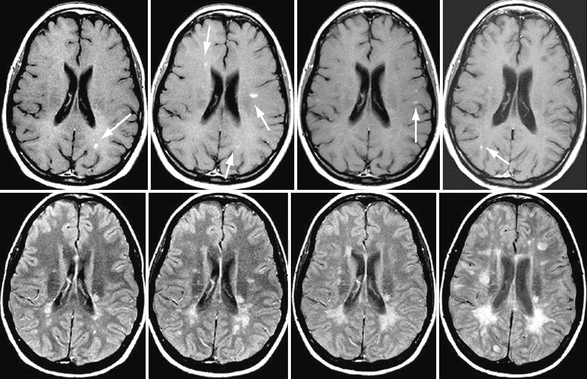
FIGURE 43-3 Relapsing form of MS. Serial, CE T1W (top row) and FLAIR (bottom row) MR images of the brain obtained yearly in a patient with a typical relapsing form of MS and progressive disability. Note the new lesions that appeared during this 3-year follow-up, some of them showing gadolinium enhancement (arrows).
(From Cañellas AR, Gols AR, Izquierdo JR, et al. Idiopathic inflammatory-demyelinating diseases of the central nervous system. Neuroradiology 2007; 49:393-469.)
Multiple Sclerosis Variants
Marburg Disease
In Marburg disease, MRI typically shows multiple focal T2 lesions of varying size, which may coalesce to form large white matter plaques disseminated throughout the hemispheric white matter and brain stem (Fig. 43-4). Perilesional edema is often present. The lesions may show enhancement. A similar imaging pattern is also seen in ADEM.
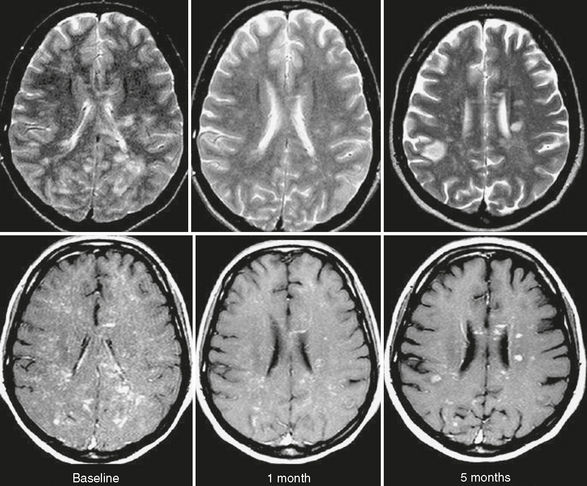
FIGURE 43-4 Marburg disease. Serial T2W (top row) and contrast-enhanced T1W (bottom row) MR images of the brain obtained in a patient with a final diagnosis of fulminant IIDD. Note multiple contrast-enhanced focal lesions diffusely involving the cerebral white matter. Some of the lesions are persistent, whereas others are new. The patient died 5 months after symptom onset.
(From Cañellas AR, Gols AR, Izquierdo JR, et al. Idiopathic inflammatory-demyelinating diseases of the central nervous system. Neuroradiology 2007; 49:393-469.)
A fulminant course can also be present in acute IIDDs showing a tumefactive or Balo-like lesion.
Schilder’s Disease
Schilder’s disease is a rare acute or subacute disorder that can be defined as a specific clinicoradiologic presentation of IIDD. It commonly affects children and young adults. The clinical spectrum of Schilder’s disease includes psychiatric predominance, acute intracranial hypertension, intermittent exacerbations, and progressive deterioration. Imaging studies show large ring-enhancing lesions involving both hemispheres, sometimes symmetrically, and located preferentially in the parieto-occipital regions. These large, focal demyelinating lesions can resemble a brain tumor, an abscess, or even adrenoleukodystrophy. MR features that suggest possible Schilder’s disease include large and relatively symmetric involvement of both brain hemispheres, incomplete ring enhancement, minimal mass effect, restricted diffusivity, and sparing of the brain stem (Fig. 43-5).
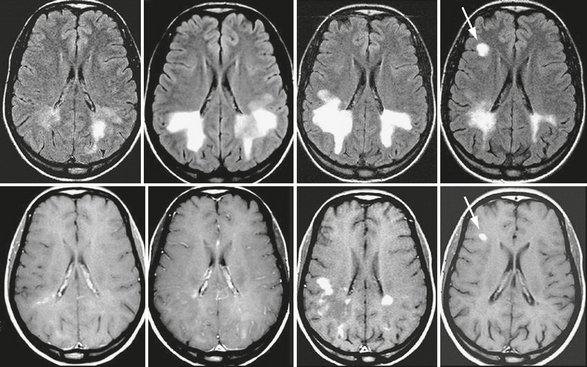
FIGURE 43-5 Schilder’s disease. Serial brain MR images in a patient with Schilder’s disease who later developed clinically definite MS. Transverse fast-FLAIR images (top row) and contrast-enhanced T1W (bottom row) images obtained serially over 6 months. Note the progressive appearance of large, bilateral, almost symmetric lesions in the posterior periventricular white matter. Despite considerable extension of the lesions there is no mass effect. The 6-month scan obtained during an episode of optic neuritis shows a new contrast-enhancing lesion in the right frontal white matter (arrow). A final diagnosis of relapsing-remitting MS was established.
(From Cañellas AR, Gols AR, Izquierdo JR, et al. Idiopathic inflammatory-demyelinating diseases of the central nervous system. Neuroradiology 2007; 49:393-469.)
Histopathologically, Schilder’s disease consistently shows well-demarcated demyelination and reactive gliosis with relative sparing of the axons. Microcystic changes and even frank cavitation can occur. The clinical and imaging findings usually show a dramatic response to corticosteroids.
Balo’s Concentric Sclerosis
Balo’s concentric sclerosis is thought to be a rare, aggressive variant of MS that can lead to death in weeks to months. The pathologic hallmarks of the disease are large demyelinated lesions showing a peculiar pattern of alternating layers of preserved and destroyed myelin. One possible explanation for the concentric alternating bands in this variant of MS may be that sublethal tissue injury is induced at the edge of the expanding lesion, which would then stimulate the expression of neuroprotective proteins to protect the rim of periplaque tissue from damage, thereby resulting in alternative layers of preserved and nonpreserved myelinated tissue.7
These alternating bands can be identified on T2W MRI, which typically shows concentric hyperintense bands corresponding to areas of demyelination and gliosis alternating with isointense bands corresponding to normal myelinated white matter (Figs. 43-6 and 43-7). This pattern can appear as multiple concentric layers (onionskin lesion), as a mosaic, or as a “floral” configuration. The center of the lesion usually shows no layering because of massive demyelination. Contrast enhancement and decreased diffusivity are frequent in the outer rings (inflammatory edge) of the lesion (see Fig. 43-7). On MRI this Balo pattern can be isolated, multiple, or mixed with typical MS-like lesions.
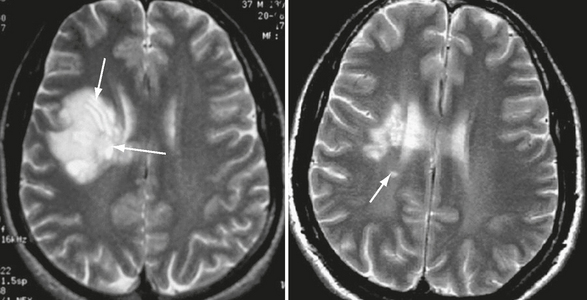
FIGURE 43-6 Balo-like lesion in a patient who converted to MS. Transverse T2W MR image of the brain shows a large focal lesion within the right frontal white matter. The striking lamellated pattern of alternating bands of demyelination and relatively normal white matter, reflecting either spared or remyelinated regions, is clear in this image (arrows, left). Note partial resolution of the large hemispheric lesion in a follow-up T2W MR image obtained 4 years after symptom onset and the presence of a new lesion (arrow, right).
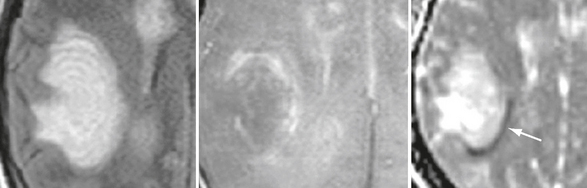
FIGURE 43-7 Balo-like lesion in patients with acute disseminated encephalomyelitis. Transverse T2W (left) and contrast-enhanced T1W (middle) MR images and apparent diffusion coefficient (ADC) map (right). Observe the alternating concentric bands, peripheral contrast uptake, and decreased peripheral diffusivity (arrow).
(From Cañellas AR, Gols AR, Izquierdo JR, et al. Idiopathic inflammatory-demyelinating diseases of the central nervous system. Neuroradiology 2007; 49:393-469.)
Tumefactive or Pseudotumoral IIDDs
In some cases, pseudotumoral IIDDs are the first clinical and radiologic manifestation of MS. More commonly, tumefactive demyelinating plaques affect patients with a known diagnosis of MS (Fig. 43-8). In rare cases, pseudotumoral IIDDs have a relapsing course, with single or multiple pseudotumoral lesions appearing over time in different locations (Fig. 43-9).
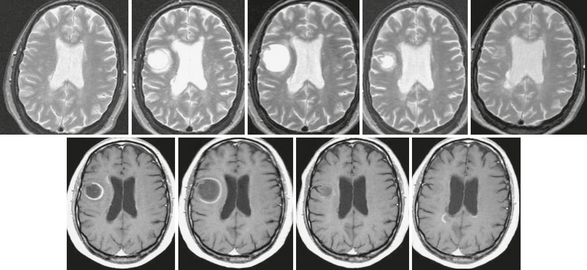
FIGURE 43-8 Tumefactive form of relapsing-remitting MS. T2W (top row) and contrast-enhanced T1W (bottom row) serial MR images of the brain acquired over 12 months in a patient with the relapsing-remitting form of MS. Note the initial increase and later decrease in size of the right frontal lobe pseudotumoral lesion, which is almost imperceptible on the 12-month scan. The lesion shows an open ring–enhancing pattern of contrast uptake, with the open margin facing the gray matter. The pseudotumoral lesion was asymptomatic.
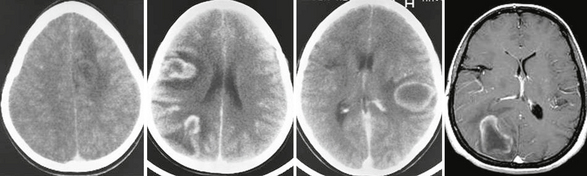
FIGURE 43-9 Tumefactive relapsing course. Serial contrast-enhanced CT and T1W MR images obtained in a 10-year-old girl who experienced several acute relapses over a period of several years related to pseudotumoral bihemispheric lesions.
(From Cañellas AR, Gols AR, Izquierdo JR, et al. Idiopathic inflammatory-demyelinating diseases of the central nervous system. Neuroradiology 2007; 49:393-469.)
On CT or MRI the pseudotumoral plaques usually present as large, single or multiple focal lesions within the cerebral hemispheres. Clues that can help to differentiate these lesions from a brain tumor are the relatively minor mass effect and the presence of incomplete ring-enhancement on gadolinium-enhanced T1-weighted (T1W) images, with the open border facing the gray matter of the cortex or basal ganglia (Fig. 43-10),8 sometimes associated with a rim of peripheral hypointensity on T2W sequences.
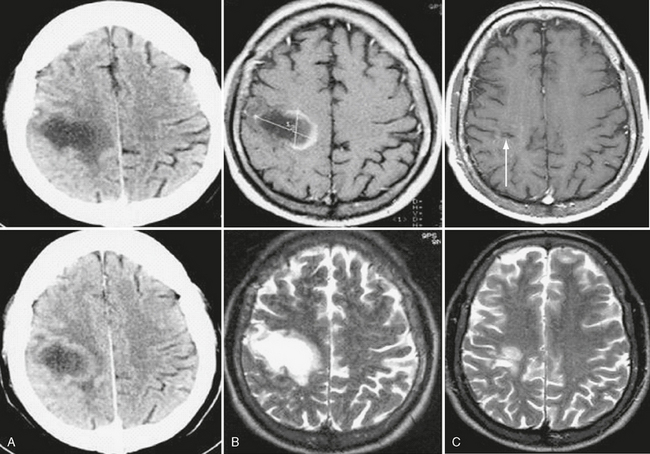
FIGURE 43-10 Tumefactive inflammatory demyelinating lesion. Unenhanced and contrast-enhanced CT (A) and T2W and contrast-enhanced T1W MR images (B) of brain show a posterior frontal lesion with minimal surrounding vasogenic edema and no mass effect. Observe the ring-enhancing pattern of contrast agent uptake, with the open margin facing the cortical gray matter. A follow-up brain MR image performed 1 year later (C) shows almost complete resolution of the lesion. The necrotic focus (arrow) in the subcortical white matter corresponds to the site of a brain biopsy, which confirmed the diagnosis of inflammatory demyelinating lesion.
Diagnosis
These principles were codified in 1983 by the Poser committee,9 which specified that the diagnosis of clinically definite MS could be based on either (1) occurrence of two attacks and clinical evidence of two lesions or (2) two attacks and clinical evidence of one lesion plus paraclinical evidence of a second lesion. MRI was not considered at that time because it was then a new, untested technique.
In 2001, new diagnostic criteria were published that incorporated a precisely defined role for MRI.10 These 2001 McDonald criteria integrated MRI criteria into their scheme to demonstrate dissemination of demyelinating lesions in space (Table 43-2). To demonstrate this principle, brain MR images must meet the Barkhof-Tintoré criteria, in which a threshold of at least three of the following four features must be achieved: (1) one gadolinium-enhancing lesion or nine T2 hyperintense lesions if gadolinium-enhancing lesions are not present, (2) at least one infratentorial lesion, (3) at least one juxtacortical lesion, and (4) at least three periventricular lesions. When three of these four parameters are not fulfilled, the presence of two or more subclinical lesions consistent with MS on brain MRI plus CSF detection of oligoclonal bands or a raised IgG index are required to demonstrate DIS. DIT can be demonstrated with MRI when new lesions have developed at least 3 months after the clinical onset. This can be a gadolinium-enhancing lesion on a scan obtained more than 3 months after a clinically isolated syndrome if it is not associated with the initial clinical event. When the scan shows no new enhancing lesion, a subsequent scan is required to demonstrate new T2 and/or gadolinium-enhancing lesions. This criterion requires between one and three MR images to be performed, with a minimum delay between symptom onset and the first scan of at least 3 months.
TABLE 43-2 MRI Criteria of Dissemination in Space (DIS) and Dissemination in Time (DIT) for Diagnosing MS

In 2005, a consensus revision of the McDonald criteria based on new evidence was proposed (see Table 43-2).13 Three major changes were introduced in the 2005 McDonald criteria. The first modification attempted to simplify the criteria for DIT. This was achieved by establishing the simple condition of demonstrating a new T2 lesion on a brain MR image at any time as compared with a reference scan done at least 30 days after the onset of the initial clinical event. The reason for selecting the 30-day time period was to exclude new T2 lesions occurring in the first few weeks after the onset of the first clinical episode, which would not be considered a new separate event. The second change proposed endeavored to better define the role of MRI of spinal cord lesions for demonstrating DIS.
A new version of the McDonald criteria (McDonald 2010)6 proposed new MR imaging criteria, in which demonstration of DIS requires the presence of at least one asymptomatic T2 lesion in two of the four locations defined as characteristic for MS: juxtacortical, periventricular, infratentorial, and spinal cord, while demonstration of DIT requires demonstration of a new T2 lesion on a follow-up scan irrespective of the timing of the baseline scan, or concomitant enhancing and non enhancing lesions on a scan performed at any time after symptoms onset11,12 (see Table 41-2). These criteria have shown similar (high) specificity for clinically definite MS and higher sensitivity when compared with the 2001 and 2005 McDonald criteria.
Pathophysiology
Type I is distinguished by demyelination and macrophage-related products.
Type II is characterized by the presence of immunoglobulin and complement, which likely explains the good response to plasma exchange in patients with this pattern.
Type III lacks immunoglobulin and complement, yet it shows early loss of myelin-associated glycoprotein and no remyelination; in this type, demyelination has been attributed to oligodendrocyte dysfunction.
Type IV is distinguished by apoptosis of oligodendrocytes through DNA fragmentation.
In individual patients, this pathogenic heterogeneity disappears, because there is a striking homogeneity among lesions. For this reason it has been suggested that MS may represent a common name for different pathologic features.13 This hypothesis has important therapeutic implications because specific therapies can be targeted to specific mechanisms.
Pathology
The Active MS Lesion
Active inflammatory demyelinating MS plaques are typically restricted to the white matter and are characterized by a mixture of lipid-laden macrophages and large reactive astrocytes, accompanied by varying perivascular inflammation. The affected areas show marked pallor of myelin staining with relative axonal preservation, although in areas where the damage is most severe the axons can be lost, be fragmented, or display irregular tortuous and clubbed profiles. Many macrophages become engorged with phagocytosed myelin remnants and debris. The so-called granular mitosis or Creutzfeldt-Peters cell is an unusual finding in reactive astrocytes in demyelination and other reactive processes. These structures have the appearance of large chromosomes arranged like mitotic figures but are actually small fragments of the nucleus. The Creutzfeldt-Peters cells may also suggest a diagnosis of glioma; however, the even distribution of the cells, the associated presence of large numbers of infiltrating macrophages in the setting of myelin loss, and relative axonal preservation should confirm that it is an inflammatory demyelinating disease and exclude the diagnosis of glioma.
Imaging
CT
Contrast-enhanced CT may be the first imaging procedure used in patients with tumefactive pseudotumoral inflammatory-demyelinating brain lesions, because the clinical features may suggest a brain tumor. A finding of incomplete ring enhancement with the open margin abutting the gray matter should suggest the correct diagnosis,8 because such findings are unusual for brain tumors and abscesses (see Fig. 43-10). Minimal or absent mass effect and a compatible clinical setting (young female, no history of cancer or brain infection) are additional clues.
MRI
Conventional MRI Techniques
Brain
MRI is the most sensitive imaging technique for detecting MS plaques throughout the brain and spinal cord. Proton density (PD)-weighted (PDW) or T2W MR images show areas of high signal intensity in the periventricular white matter in 98% of MS patients. MS plaques are generally round to ovoid and range from a few millimeters to more than 1 cm in diameter. They are typically discrete and focal at the early stages of the disease but become confluent as the disease progresses, particularly in the posterior hemispheric periventricular white matter (see Fig. 43-3). MS plaques tend to affect the deep white matter rather than the subcortical white matter, whereas small vessel ischemic lesions tend to involve the subcortical white matter more than the periventricular white matter. The total T2 lesion volume of the brain increases by 5% to 10% each year in the relapsing forms of MS.14
Both acute and chronic MS plaques appear bright on PDW and T2W sequences, reflecting their increased tissue water content. The signal increase indicates edema, inflammation, demyelination, reactive gliosis, and/or axonal loss in proportions that differ from lesion to lesion. The vast majority of MS patients have at least one ovoid periventricular lesion, whose major axis is oriented perpendicular to the outer surface of the lateral ventricles (Fig. 43-11). The ovoid shape and perpendicular orientation derive from the perivenular location of the demyelinating plaques (Dawson’s fingers).
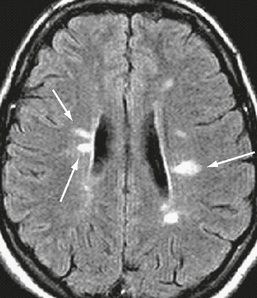
FIGURE 43-11 Relapsing-remitting MS. Transverse fast-FLAIR MR image shows typical ovoid demyelinating plaques (arrows), whose major axis is perpendicular to the ventricular wall.
MS lesions tend to affect specific regions of the brain, including the periventricular white matter situated superolateral to the lateral angles of the ventricles, the callososeptal interface along the inferior surface of the corpus callosum, the corticojuxtacortical regions, and the infratentorial regions. Focal involvement of the periventricular white matter in the anterior temporal lobes is typical for MS and rarely seen in other white matter disorders (Fig. 43-12). The lesions commonly found at the callososeptal interface are best depicted by sagittal fast-FLAIR images; so this sequence is highly recommended for diagnostic MRI studies (Fig. 43-13).