Neoplasms of the Spine
A. JAMES BARKOVICH
SPINAL TUMORS—INTRODUCTION
Tumors of the spinal cord in children are slowly growing masses with clinical manifestations that are often subtle and slowly progressive. As a result, in the past their diagnosis was somewhat delayed and occurred late in the course of the disease. The widespread use of magnetic resonance imaging (MRI) has greatly facilitated the diagnosis of spinal tumors, allowing earlier detection and therapy.
Although the neoplasms that involve the pediatric spinal cord are similar to those affecting adults, the incidence and the presentation of the various tumors are often quite different. In this chapter, the epidemiology, clinical manifestations, and pathologic/radiologic manifestations of intraspinal neoplasms in children are discussed.
GENERAL IMAGING CHARACTERISTICS OF SPINAL TUMORS
The introductory section of Chapter 7 discussed general characteristics of brain tumors. In that discussion, the importance of determining whether a tumor is located within the brain (intraparenchymal) or outside of the brain (extraparenchymal) was stressed. A similar distinction must be made in tumors of the spine: does the tumor originate within the spinal cord (intramedullary tumor) or outside of the spinal cord (extramedullary tumor)?
Differentiating an intramedullary mass from an extramedullary mass by imaging is usually straightforward. Intramedullary masses cause enlargement of the spinal cord as viewed in the sagittal, coronal, and axial planes. The spinal cord above and below the mass remains in its normal location within the spinal canal. The margins of the intramedullary mass may be sharply defined (particularly in congenital tumors, such as dermoids and epidermoids) or indistinct (particularly in infiltrating astrocytomas). Extramedullary masses always appear sharply marginated in at least one imaging plane. When small, they are usually clearly separated from the cord by cerebrospinal fluid (CSF). When large, the CSF space between the mass and the cord is obliterated. In addition, the mass may compress the cord. If compressed by a laterally located mass, the cord will appear enlarged if viewed in the sagittal plane; therefore, coronal and axial planes are necessary to make the distinction. If compressed by an anteriorly or a posteriorly located mass, the cord will appear enlarged in the coronal plane; imaging in the sagittal and axial planes will allow diagnosis. Sometimes, the site of tumor origin is clarified after tumor enhancement by intravenous administration of contrast.
A potential pitfall in the diagnosis of intramedullary tumors is in the differentiation of tumors from nonneoplastic processes in the spinal cord. In children, the major problem is in the differentiation of tumors from foci of demyelination. For example, children with acute disseminated encephalomyelitis or multiple sclerosis (see Chapter 3) can present with signs and symptoms referable to a spinal cord lesion. In either disease, a region of focal cord enlargement with prolongation of T2 relaxation time may be identified. Demyelinating plaques may be enhanced after the administration of contrast. Perfusion imaging, which helps in this differentiation in brain tumors (see Chapter 7), is not yet practical in the spine because of susceptibility effects from the spinal column and the small size of the spinal canal. Therefore, demyelination is most easily differentiated from tumor by assessing the shape and size of the lesion at the time of presentation. Neoplasms of the spine are round to ovoid in shape and often have associated cysts (Fig. 10-1A and B). They typically cause considerable enlargement in the cord diameter at the time of presentation because they rarely cause symptoms when they are less than 2 cm in diameter. Plaques of demyelination are usually flame shaped and rarely cause significant enlargement of the spinal cord (Fig. 10-1C and D). Associated cysts are almost never present. It may be helpful to image the brain to look for other characteristic foci of demyelination (see Chapter 3); if present, they establish the diagnosis. If no other lesions are found, and uncertainty persists as to whether an intramedullary lesion is tumor or plaque, a follow-up MR exam with and without paramagnetic contrast should be obtained 4 to 6 weeks after the initial exam. Tumor will be unchanged or larger, whereas a focus of demyelination should be smaller and the enhancement characteristics should be different than on the first study. A delay of 6 weeks will not impact on the outcome of a patient with a tumor, but biopsy of an acute demyelinating plaque may leave the patient with a worse neurological deficit.
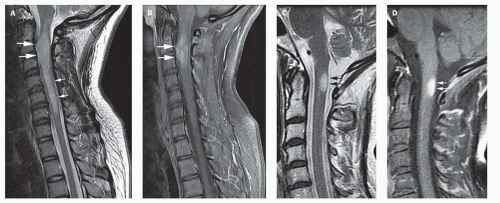 FIG. 10-1. Cervical spinal cord astrocytoma; comparison with demyelinating plaque. A. Sagittal T2-weighted image shows hyperintensity within the cord extending from the foramen magnum to the bottom of C-6. At C-2 to C-3, the lesion is ovoid and the cord is expanded (large white arrows), strongly suggesting the presence of tumor. The less expanded portion (small white arrows) probably represents edema or, possibly, a “presyrinx”. B. Contrast-enhanced sagittal T1-weighted image shows heterogeneous enhancement (white arrows) of the ovoid, expanded region of the cord. C and D. Sagittal T2-weighted image (C) and contrast-enhanced T1-weighted image (D) show a demyelinating plaque. The lesion (arrows) is flame shaped and causes minimal mass effect. |
INTRAMEDULLARY TUMORS
Clinical Presentation
Intramedullary (intrinsic) neoplasms of the spinal cord comprise 6% to 10% of neoplasms of the central nervous system in children (1,2,3,4,5,6) and approximately one third of spinal neoplasms of childhood (4,7,8,9). All age groups are affected; however, intramedullary tumors are most frequently seen in children toward the end of the first decade and beginning of the second decade of life. Males and females are equally affected (2,3,8,10). Spinal neoplasms are relatively less common in children than in adults; the ratio of intracranial to intraspinal neoplasms in children varies between 10:1 and 20:1 (as compared to 5:1 in adults) (3).
In the past, patients with spinal cord neoplasms generally presented for treatment months, or even years, after the onset of clinical symptoms (3,8,10,11,12). The availability of MR has resulted in earlier diagnosis; nonetheless, the patient’s course will often have been punctuated by exacerbations and remissions of symptoms, possibly due to alteration in the edema surrounding the neoplasm (9,10,11). Weakness with numbness or pain is the most common presenting complaint (3,13). The most common type of pain, occurring in approximately 70% of patients, is spinal pain. This pain is dull and aching in quality and is localized to bone segments adjacent to the tumor (3). The cause of this pain is unknown but may result from distention of the dural tube by the swollen spinal cord (3). Sometimes children will complain of nocturnal pain that awakens them from sleep (9). The second most common type of pain seen in children is root pain, which is indistinguishable from pain caused by compression of a nerve root by an extramedullary process (3). Finally, some patients manifest tract pain, a vague, burning type of pain associated with paresthesias. The burning and paresthesias typically occur caudal to the tumor and are believed to result from infiltration of the lateral spinothalamic tracts by tumor (3,10,14).
Children of different age groups often present with very different symptomatology. Young children and infants often have severe pain, motor regression, weakness, or frequent falls (4,9,10,11,15), whereas older children more commonly present with clumsiness, progressive scoliosis (thoracic tumors), torticollis (cervical tumors), gait disturbance, sphincter disturbance, or spinal deformity (2,3,6,11,13,16). Weakness of the extremities is a common finding in all patients. In tumors of the cervical spinal cord, the most common location in children, upper extremity weakness may or may not be accompanied by weakness of the lower extremities, neck pain, muscle atrophy, dysesthesias, or hyperreflexia (6). Although sensory levels may be present, they are less common in intramedullary than in extramedullary tumors. Sphincter dysfunction, when present, may not occur until late in the course of the illness (10,11,15); early presentation may indicate a high-grade (fastgrowing) tumor (17).
Up to 15% of patients with spinal cord tumors may present with symptoms of increased intracranial pressure, most likely a result of either greatly elevated CSF protein content, which blocks the normal CSF pathways and impairs CSF resorption, or blockage of the foramen magnum by cervicomedullary tumors (see Chapter 8) (13,18,19,20,21). The increased intracranial pressure does not misdirect the diagnostic workup if the patients have signs of an intraspinal process on physical exam. However, patients having no signs or symptoms of a spinal process may suffer from a considerable delay in diagnosis.
Pathology
The most common histologic type of spinal cord tumor in children is astrocytoma (60%), followed by ependymoma (15%-30%) (5,9,13,22,23,24). Thus, the relative incidence of intramedullary spinal cord tumors in children is quite different from that in adults, in whom more than half of intramedullary tumors are ependymomas. Another difference from adults is that the myxopapillary ependymoma, a benign (WHO Grade I) form of ependymoma that comprises 40% to 50% of spinal ependymomas in adults, is rare in children, comprising only 8% to 12% of spinal ependymomas in childhood (25,26); most pediatric ependymomas appear to be WHO Grade II (cellular, papillary, epithelial, or clear cell). Malignant astrocytomas and glioblastomas of the cord are relatively more common in children than in adults, comprising as many as 25% of pediatric astroglial spinal cord tumors (9,27).
Other intramedullary tumors may develop in children, including gangliogliomas and gangliocytomas (28,29), germinomas (5,23,30,31), primitive neuroectodermal tumors (PNETs) (32,33), atypical teratoid/ rhabdoid tumors (17,34,35,36,37), oligodendrogliomas (38), pleomorphic xanthoastrocytomas (23,39), melanocytomas (23), teratomas (40), and Langerhans cell histiocytosis (5,41). Very rarely, intramedullary tumors may be metastases from fourth ventricular tumors (that presumably seed via the obex into the central canal of the spinal cord) (42).
Other intramedullary tumors may develop in children, including gangliogliomas and gangliocytomas (28,29), germinomas (5,23,30,31), primitive neuroectodermal tumors (PNETs) (32,33), atypical teratoid/ rhabdoid tumors (17,34,35,36,37), oligodendrogliomas (38), pleomorphic xanthoastrocytomas (23,39), melanocytomas (23), teratomas (40), and Langerhans cell histiocytosis (5,41). Very rarely, intramedullary tumors may be metastases from fourth ventricular tumors (that presumably seed via the obex into the central canal of the spinal cord) (42).
Just as with their intracranial counterparts, the extent of resection of spinal tumors is the primary determinant of the rate of recurrence and the expected length of survival (43). Astrocytomas are generally more difficult to completely resect than ependymomas; thus, patients with astrocytomas have poorer postoperative courses and shorter survival (13,44,45). Histology of the tumor is also an important factor in determining expected survival. In one study, 10-year survival for pilocytic astrocytomas of the spinal cord was 81% as compared to 15% for diffuse fibrillary astrocytomas (46) and even worse for glioblastoma multiforme, where subarachnoid spread was found in up to 60% of cases (47). Among patients with fibrillary astrocytomas, the grade of the tumor was the most significant predictor of survival (46). Among ependymomas, too, high tumor grade (but not histology—there does not appear to be any difference in the biologic behavior of the different histologic types of WHO Grade II ependymomas [5]) was the most significant variable for prediction of dissemination through the cerebrospinal axis (48); dissemination is a grave prognostic factor.
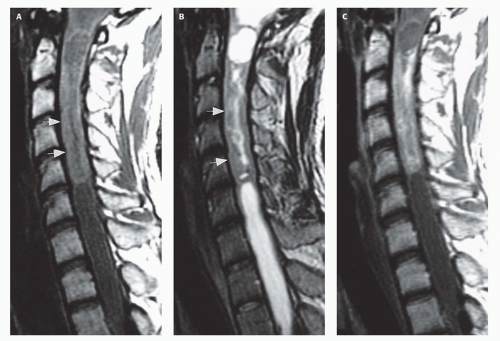 FIG. 10-2. Cervical spinal cord astrocytoma with associated syrinx. A. Sagittal T1-weighted image shows expansion of the spinal cord from the foramen magnum to the T2 level. The relatively high signal intensity of the solid tumor in the lower cervical spinal cord (arrows) contrasts with T1 prolongation of cystic regions extending into the expanded brainstem and thoracic spinal cord. B. Sagittal T2-weighted image shows that the central solid portion of the tumor (arrows) is of heterogeneous low signal intensity compared with the marked hyperintensity of the cysts in the brainstem and thoracic cord. C. Postcontrast T1-weighted image shows mild enhancement of the solid portion of the tumor. |
Intramedullary neoplasms tend to be located more rostrally in children than in adults (1,3). Nearly 50% of intramedullary tumors in children are cervical or cervicothoracic, as compared with 28% in adults. Intramedullary tumors in children usually occupy a small number of cord segments; however, the extent of cord involvement is often increased by the presence of cysts rostral or caudal to the tumor. Such peritumoral cysts have been reported in 40% of spinal cord astrocytomas (49) and in up to 80% of ependymomas (50,51).
Imaging
Magnetic resonance is the modality of choice for imaging patients with suspected or known intramedullary neoplasms (52,53,54,55,56). MR of most tumors will show an enlarged spinal cord with T1 hypointensity and T2/FLAIR hyperintensity at the location of the tumor (Figs. 10-1 to 10-7). As in the brain, tumors with high cell density, such as PNETs and atypical teratoid/rhabdoid tumors, have relatively little free water and therefore show less T2 hyperintensity (32,34,37), although this characteristic seems less reliable in the spinal cord than in the brain (57). The regions of abnormal T1 and T2 signal within the expanded cord may represent tumor, necrosis, cyst, or “presyrinx” (Fig. 10-1) (58); therefore, intravenous administration of paramagnetic contrast (Figs. 10-1, 10-2, and 10-5 to 10-7) is essential. Contrast enhancement is usually nodular, heterogeneous (Figs. 10-2, 10-4, and 10-5), or ring-like (Fig. 10-7); although there are exceptions (59,60), enhancement is seen
in the large majority of intramedullary spinal neoplasms in children (22,25,26,28,31,41,50,51,61,62). The region of contrast enhancement corresponds to the solid portion of the tumor and usually assists in distinguishing tumor (which typically enhances) from cyst and necrosis (which do not enhance, although contrast may leak into a cyst lined by tumor as in Fig. 10-5). Astrocytomas (Figs. 10-1 and 10-2) are usually located more eccentrically in the spinal cord, enhance less intensely or not at all, and the enhancement is less sharply demarcated from the nonenhancing tissue than in ependymomas (Figs. 10-4 and 10-5) (63). In addition, the tumor margins of astrocytomas usually extend beyond the enhancing tissue. Ependymomas are usually located centrally within the spinal cord; the margin of tumor enhancement is sharper and corresponds to the margin of solid tumor (25,26,61). Germinomas and gangliogliomas (Figs. 10-6 and 10-7) are typically sharply circumscribed, as well. Heterogeneity of the tumor on T1-weighted images is said to be suggestive of ganglioglioma (Fig. 10-6) (50), although astrocytomas may have a similar appearance (Fig. 10-1). A finding of T2 hypointensity at the superior and inferior borders of the mass (Fig. 10-5), indicating prior hemorrhage, is suggestive of ependymomas (64); however, this sign is seen in only about 20% of ependymomas, (51). No specific MR characteristics allow differentiation of benign from malignant spinal cord tumors in children, although the presence of leptomeningeal spread at the time of presentation suggests malignancy (65). In practice, preoperative differentiation of these tumors is of little clinical importance because the operative approach does not differ. It is much more important to determine whether the tumor is intramedullary or extramedullary and to accurately identify the level of the solid portion of the tumor.
in the large majority of intramedullary spinal neoplasms in children (22,25,26,28,31,41,50,51,61,62). The region of contrast enhancement corresponds to the solid portion of the tumor and usually assists in distinguishing tumor (which typically enhances) from cyst and necrosis (which do not enhance, although contrast may leak into a cyst lined by tumor as in Fig. 10-5). Astrocytomas (Figs. 10-1 and 10-2) are usually located more eccentrically in the spinal cord, enhance less intensely or not at all, and the enhancement is less sharply demarcated from the nonenhancing tissue than in ependymomas (Figs. 10-4 and 10-5) (63). In addition, the tumor margins of astrocytomas usually extend beyond the enhancing tissue. Ependymomas are usually located centrally within the spinal cord; the margin of tumor enhancement is sharper and corresponds to the margin of solid tumor (25,26,61). Germinomas and gangliogliomas (Figs. 10-6 and 10-7) are typically sharply circumscribed, as well. Heterogeneity of the tumor on T1-weighted images is said to be suggestive of ganglioglioma (Fig. 10-6) (50), although astrocytomas may have a similar appearance (Fig. 10-1). A finding of T2 hypointensity at the superior and inferior borders of the mass (Fig. 10-5), indicating prior hemorrhage, is suggestive of ependymomas (64); however, this sign is seen in only about 20% of ependymomas, (51). No specific MR characteristics allow differentiation of benign from malignant spinal cord tumors in children, although the presence of leptomeningeal spread at the time of presentation suggests malignancy (65). In practice, preoperative differentiation of these tumors is of little clinical importance because the operative approach does not differ. It is much more important to determine whether the tumor is intramedullary or extramedullary and to accurately identify the level of the solid portion of the tumor.
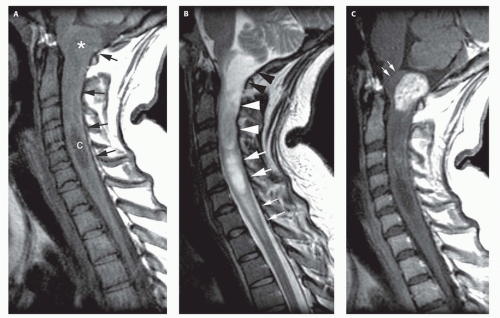 FIG. 10-3. Cervicomedullary astrocytoma. A. Sagittal T1-weighted image shows a partially cystic mass (black arrows) in the mid and upper cervical spinal cord. A frank cyst (c) is present from C-4 to C-6. A dorsally exophytic component (white asterisk) is seen at the craniocervical junction. B. Sagittal T2-weighted image shows multiple components in the cervical spinal cord. The lowest component (small white arrows) likely represents vasogenic edema. The next most rostral component, of high signal intensity, (large white arrows) likely represents a tumor-associated cyst. The next component (large white arrowheads) likely represents solid tumor with high cellularity. The most rostral component (large black arrowheads) likely represents solid tumor with high water content/low cellularity. C. Postcontrast sagittal T1-weighted image shows minimal enhancement of the more cellular solid component of the tumor at C-2 to C-3 and marked enhancement of the dorsally exophytic component near the cervicomedullary junction. The white arrows point to the displaced cervicomedullary junction. This marks the presumed level of the pyramidal decussation. High-grade tumors break through the horizontally oriented axons at this level to invade the medulla, whereas lower grade tumors displace the decussation and expand dorsally, as in this case. |
In 20% to 40% of patients, cysts are present caudal or rostral to the solid portion of the tumor (Figs. 10-1, 10-5, and 10-6) (3,49,53); this number may be as high as 80% in ependymomas (50,51,66). These cysts may result from secretion of fluid by the tumor or by obstruction of normal CSF pathways within the cord and subarachnoid space; the latter should be considered syringomyelia. The solid portion of the tumor often cannot be differentiated from the cyst without the intravenous administration of a contrast (Fig. 10-5). Axial and sagittal T1-weighted sequences may be of use in defining the full extent of the associated cyst. The full extent of the cyst is not critical information, however, because it usually resolves after resection of the tumor and elimination of the obstruction.
The differentiation of tumor-associated syringomyelia from congenital or posttraumatic syringomyelia is difficult, even impossible,
without the use of contrast-enhanced MR. Edema with reactive astrogliosis, which has the same signal characteristics as tumor (T1 hypointensity and T2 hyperintensity), is frequently present at the caudal and rostral ends of benign syrinx cavities (see Chapter 9) (67,68); this finding may be a “presyrinx,” which is slowly evolving into true syringohydromyelia (58). Although the presence of anomalies of the craniocervical junction such as cerebellar tonsillar ectopia, occipitalization of the atlas, and the Klippel-Feil anomaly suggest that a syrinx is nonneoplastic, these anomalies can occur in conjunction with spinal cord tumors as well (67,69). Moreover, the expansion of the intramedullary tumor that obstructs CSF flow in the spinal subarachnoid space can cause a syringohydromyelia (Figs. 10-2 and 10-5) identical to that seen associated with tonsillar ectopia (67,68). Patients with typical syringohydromyelia (see Chapter 9) and an obvious cause for the syrinx, such as a Chiari I malformation, do not need a postcontrast MR sequence. However, if any question exists regarding the cause of the syrinx, paramagnetic contrast should be administered to look for enhancing tumor. If an area of contrast enhancement is present within or adjacent to the syrinx, the diagnosis of tumor-related cyst should be strongly considered. Rarely, an enhancing inflammatory mass can cause cavitary cord disease.
without the use of contrast-enhanced MR. Edema with reactive astrogliosis, which has the same signal characteristics as tumor (T1 hypointensity and T2 hyperintensity), is frequently present at the caudal and rostral ends of benign syrinx cavities (see Chapter 9) (67,68); this finding may be a “presyrinx,” which is slowly evolving into true syringohydromyelia (58). Although the presence of anomalies of the craniocervical junction such as cerebellar tonsillar ectopia, occipitalization of the atlas, and the Klippel-Feil anomaly suggest that a syrinx is nonneoplastic, these anomalies can occur in conjunction with spinal cord tumors as well (67,69). Moreover, the expansion of the intramedullary tumor that obstructs CSF flow in the spinal subarachnoid space can cause a syringohydromyelia (Figs. 10-2 and 10-5) identical to that seen associated with tonsillar ectopia (67,68). Patients with typical syringohydromyelia (see Chapter 9) and an obvious cause for the syrinx, such as a Chiari I malformation, do not need a postcontrast MR sequence. However, if any question exists regarding the cause of the syrinx, paramagnetic contrast should be administered to look for enhancing tumor. If an area of contrast enhancement is present within or adjacent to the syrinx, the diagnosis of tumor-related cyst should be strongly considered. Rarely, an enhancing inflammatory mass can cause cavitary cord disease.
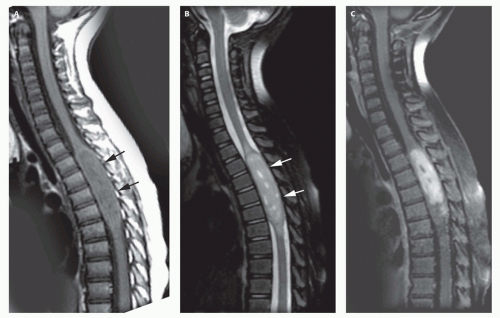 FIG. 10-4. Spinal cord ependymoma. A. Noncontrast sagittal T1-weighted image shows enlargement of the spinal cord (black arrows) with heterogeneous prolonged T1 relaxation within the upper thoracic spinal cord. B. Sagittal T2-weighted image shows that the area of widened spinal cord (white arrows) is very sharply marginated. This type of sharp demarcation is characteristic of low-grade ependymomas. Small areas of vasogenic edema are present rostral and caudal to the mass. C. Postcontrast sagittal T1-weighted image shows the tumor to enhance heterogeneously. |
On rare occasions, intramedullary tumors have disseminated throughout the subarachnoid spaces by the time of presentation (38,39,70). Although this finding is generally associated with higher tumor grade, dissemination may be found in low-grade tumors, as well; the reason for their early dissemination is uncertain (38,70,71). Such patients may initially come to medical attention because of signs of increased intracranial pressure. It is important to remember, as discussed in Chapter 8, that CSF dissemination of spinal tumors can cause hydrocephalus and that the spine should be investigated in children (or adults) with unexplained hydrocephalus which worsens despite CSF diversion.
Congenital intramedullary tumors, such as lipomas, teratomas, hamartomas, dermoids, and epidermoids, are discussed in Chapter 9.
EXTRAMEDULLARY TUMORS
Approximately two thirds of all intraspinal tumors of childhood are extramedullary; 50% are extradural and 10% to 15% are intradural (72,73). Extramedullary neoplasms can be classified by a number of different criteria; they will be classified in this chapter by their site of origin. Clinically, patients with extramedullary tumors present with signs of progressive myelopathy. Presentation is similar to intramedullary tumors in that weakness, predominantly of the lower extremities and trunk, is the most common presenting symptom. Diffuse back pain and radicular (“root”) pain are frequently present. Torticollis, upper extremity weakness, scoliosis, and urinary incontinence are other common signs and symptoms (2,14,15). The histological type of tumor cannot be predicted by its clinical presentation.
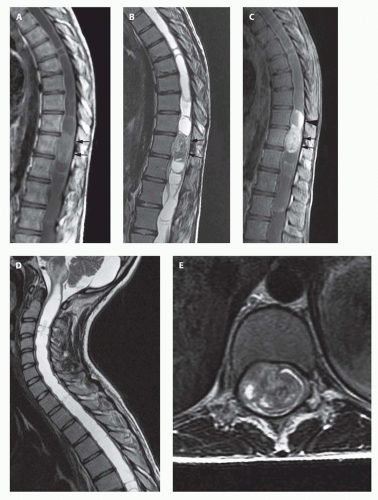 FIG. 10-5. Thoracic ependymoma with holocord syringomyelia. Sagittal T1-weighted (A) and T2-weighted (B) images show a nearly completely fluid-filled spinal cord with a solid tumor component (black arrows) in the lower thoracic region. Contrastenhanced T1-weighted image (C) shows that the solid portion of the tumor enhances (black arrows), along with the cystic portion above it (large black arrowhead), suggesting that the enhancing cyst is a tumor cyst rather than an associated syrinx. Sagittal T2-weighted image of the cervicothoracic spine (D) shows that the syrinx extends into the medulla (syringobulbia). Axial T2-weighted image (E) shows the tumor to be extremely heterogeneous with cysts (hyperintense) and mineralization/hemorrhage (hypointense). |
Cerebrospinal Fluid Dissemination of Intracranial Neoplasms
The dissemination of tumor from intracranial neoplasms via the CSF occurs more frequently in the pediatric age group than in adults. This manner of tumor spread is most commonly observed in medulloblastomas (74). Up to one third of patients with medulloblastoma will eventually disseminate tumor through the CSF, most often to the spine (75). CSF-borne metastases appear to be more common in children with differentiated medulloblastomas, that is, medulloblastomas with areas of glial, ependymal, and/or neuronal differentiation (76). The presence of metastases in the spinal canal indicates a much poorer prognosis for the patient (77).
Ependymomas, anaplastic gliomas, germinomas, choroid plexus tumors, and pineal parenchymal tumors (pineoblastomas and pineocytomas) also frequently disseminate through the CSF and deposit metastases in the spinal canal (39,74). Benign ependymomas uncommonly have detectable spinal metastases at the time of the initial diagnosis. Spinal deposits from ependymomas occur more frequently in anaplastic tumors or after local recurrence of a benign tumor is detected (74); fourth ventricular ependymomas spread through the CSF much more frequently than those located in the cerebrum. The fact that pineoblastomas and poorly differentiated pineocytomas metastasize to the CSF is not surprising since these neoplasms are intraventricular and, in the case of the pineoblastoma, closely resemble medulloblastomas.
Related posts:
 Techniques and Methods in Pediatric Neuroimaging
Techniques and Methods in Pediatric Neuroimaging
 Anomalies of Cerebral Vasculature: Diagnostic and Endovascular Considerations
Anomalies of Cerebral Vasculature: Diagnostic and Endovascular Considerations
 Techniques for Pediatric Neuroimaging
Techniques for Pediatric Neuroimaging
 Congenital Anomalies of the Spine
Congenital Anomalies of the Spine
 Neoplasms of the Spine
Neoplasms of the Spine
 Anomalies of the Cerebral Vasculature: Diagnostic and Endovascular Considerations
Anomalies of the Cerebral Vasculature: Diagnostic and Endovascular Considerations
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


