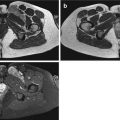
Fig. 17.1
(a, b) Nerve sheath architecture: (a) components of a nerve fiber surrounded by endoneurium. (b) Nerve trunk with five fascicles in group arrangement (Source: Penkert et al. [104])
Nerve sheath tumors are neoplasms composed of cells showing nerve sheath differentiation [74]. They represent 10–12 % of all soft tissue neoplasms [47]. The fourth edition of the WHO Classification of Tumors of Soft Tissue and Bone, published in 2013, now incorporates a chapter specifically dedicated to nerve sheath tumors [32]. Some of these tumors were previously classified under “Tumors of the Nervous System” and “Tumors of the Skin” [31, 43]. Consequently, the updated WHO classification includes several new benign and malignant nerve sheath tumor types (Table 17.1). Morton’s fibroma (neuroma) and traumatic neuroma will be discussed separately since these tumors do not belong to the group of nerve sheath tumors (Chap. 24).
Table 17.1
2013 WHO classification of nerve sheath tumors
Benign |
Schwannoma (including variants) |
Neurofibroma (including variants) |
Perineurioma |
Granular cell tumor |
Hybrid nerve sheath tumor |
Benign triton tumor |
Dermal nerve sheath myxoma |
Solitary circumscribed neuroma |
Ectopic meningioma |
Nasal glial heterotopia |
Malignant |
Malignant peripheral nerve sheath tumor |
Epithelioid malignant nerve sheath tumor |
Malignant triton tumor |
Malignant granular cell tumor |
Ectomesenchymoma |
17.2 Benign Nerve Sheath Tumors
17.2.1 Schwannomas
17.2.1.1 Epidemiology
Schwannomas represent approximately 5 % of all benign soft tissue neoplasms [47]. They can occur at all ages, though they are uncommon in children. While most literature data indicate that schwannomas are most prevalent between the ages of 20 and 50, others have observed a bimodal age distribution with peak incidences in the third and seventh decades [62]. Men and women are equally affected. Schwannomas associated with neurocutaneous syndromes tend to occur in younger patients [24].
17.2.1.2 Topography
Schwannomas most commonly involve the major nerve trunks of the head, neck, and limbs. The flexor surfaces of the extremities are more frequently affected, especially near the elbow, wrist, and knee [24, 62, 76]. Consequently, the most common topographical locations of schwannomas include the spinal roots and cervical plexus and the vagus, peroneal, and ulnar nerves. Deeply situated schwannomas are predominantly found in the posterior mediastinum and the retroperitoneum [24, 62]. Relatively few schwannomas appear on the trunk, which contrasts with the distribution of neurofibromas in neurofibromatosis type 1 (NF1). Other sites of involvement include the scalp and the hands; the feet are usually spared. Less frequently, schwannomas have been recorded in the tongue, palate, and larynx.
17.2.1.3 Histology
General Histologic Features of Schwannomas
Schwannomas are benign, slowly growing neoplasms. They originate in a nerve and are composed exclusively of Schwann cells embedded in a collagenous matrix [38, 62]. In general, they are firm, circumscribed, and encapsulated by epineurium. Unlike neurofibromas, which incorporate axons, schwannomas grow eccentrically, displacing normal elements of the nerve to one side [22]. This implies that surgical excision can usually spare the parent nerve because schwannomas are separable from the underlying nerve fibers [48].
Macroscopically, the cut surface is relatively homogeneous, tan or gray, with irregular yellow areas and cysts, particularly in large tumors. Less often schwannomas can be (partially) hemorrhagic [38]. Small schwannomas tend to be spheroid, whereas larger tumors can be ovoid, sausage-shaped, or irregularly lobulated [76].
Microscopically, two types of tissue can be distinguished in schwannomas: Antoni A and B [62]. The Antoni A tissue is composed of compactly arranged cells which are often aligned in rows and separated by clear hyaline bands, a characteristic pattern called Verocay bodies [98]. Commonly, but not invariably, a part of, or all of, a schwannoma has a less cellular Antoni B pattern in which the tumor cells are separated by a finely honeycombed eosinophilic matrix [99].
Vessels in schwannomas are usually prominent [38] and are liable to spontaneous thrombosis with consequent necrosis, and sometimes hemorrhage, in the adjacent areas. The rich vascular supply of schwannomas is reflected in the often intense enhancement of these tumors on imaging studies [76].
The histologic features of schwannomas may reflect their occasional inhomogeneous appearance on computed tomography (CT) or magnetic resonance imaging (MRI) examinations and can be summarized as follows [76]:
- 1.
Areas of decreased cellularity (Antoni B) adjacent to areas of increased cellularity (Antoni A)
- 2.
Cystic degeneration due to vascular thrombosis and subsequent necrosis
- 3.
Xanthomatous regions, containing aggregates of lipid-laden (foam) cells
Special Histologic Types of Schwannomas
Histologically, several variants of benign schwannoma can be distinguished.
Cellular schwannomas have a predominantly cellular growth but no Verocay bodies and are almost exclusively composed of Antoni A areas [62]. They appear circumscribed, if not encapsulated. They typically occur in middle-aged women and are more frequently found in deep structures, e.g., the retroperitoneum and posterior mediastinum [24]. Cellular schwannomas can cause erosion of bone and recur locally. Despite their increased cellularity, malignant transformation does not occur [34, 97].
Ancient schwannomas are long-standing schwannomas exhibiting degenerative changes including calcification, hyalinization, relative loss of Antoni A areas, and cystic cavitation [76]. Given the heterogeneity of these neoplasms and the fact that tumor nuclei may appear enlarged and hyperchromatic, differentiation with malignant soft tissue tumors may be difficult [41].
(Psammomatous) Melanotic schwannomas are characterized by a strong melanocytic differentiation of the Schwann cells. Schwann cells and melanocytes share a common precursor stem cell derived from the neural crest, hence this differentiation [44]. Melanotic schwannomas have a predilection for spinal nerve roots and commonly arise near the midline. It has been suggested that they display malignant behavior with local recurrence after surgery [24]. Over 50 % of patients suffering from melanotic schwannomas develop Carney’s syndrome, consisting of myxomas, spotty pigmentation, and endocrine overactivity producing Cushing’s syndrome [74, 75].
Plexiform (multinodular) schwannomas constitute 5 % of all schwannomas. The multinodular or plexiform pattern of growth may or may not be apparent macroscopically [24]. Only 4 % of all cases of this form reported in the literature occurred in patients with NF1; therefore, it is not to be considered associated with NF1 [33, 62, 101]. Plexiform schwannomas arise in the dermis or subcutaneous tissues and occur predominantly in young adults, without sex predilection [33, 49]. Although they can display focal nuclear pleomorphism and increased cellularity, cellular schwannomas appear to be benign lesion [76, 101].
Microcystic (reticular) schwannomas represent a new schwannoma type, recently reported in ten patients. Microcystic schwannomas are preferentially located in the gastrointestinal submucosa or subcutaneous tissue [56].
17.2.1.4 Clinical Presentation
Small schwannomas are usually asymptomatic. Pain, paresthesias, muscle atrophy, or other symptoms can occur when the tumor reaches sufficient size to compress the involved nerve or adjacent structures. On physical examination, they can be moved from side to side, but not along the long axis of the nerve [38].
17.2.1.5 Imaging Characteristics
Plain Radiography
Schwannomas are usually not seen on plain radiography, unless they are very large [7]. Sometimes they exhibit a slightly lower attenuation of the X-ray beam than muscle (Fig. 17.2). Pressure erosions on bone can be seen as widening of the intervertebral foramina or rib notching, especially in cellular schwannomas (Figs. 17.3 and 17.4) [76]. A posterior mediastinal mass on a chest radiograph should suggest the possibility of a neurogenic tumor, for they account for 30 % of posterior mediastinal mass lesions.
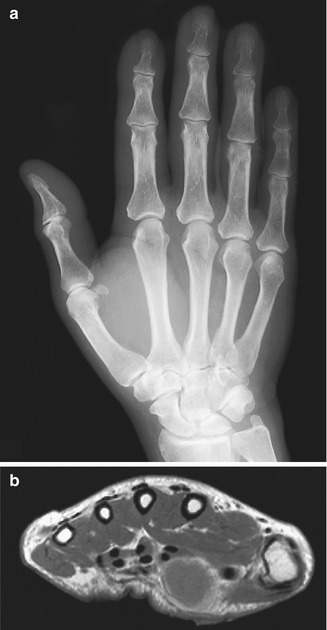
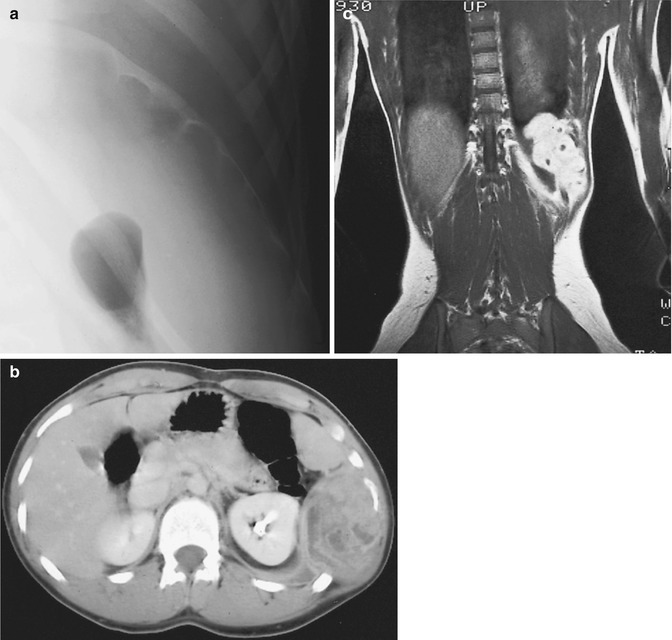
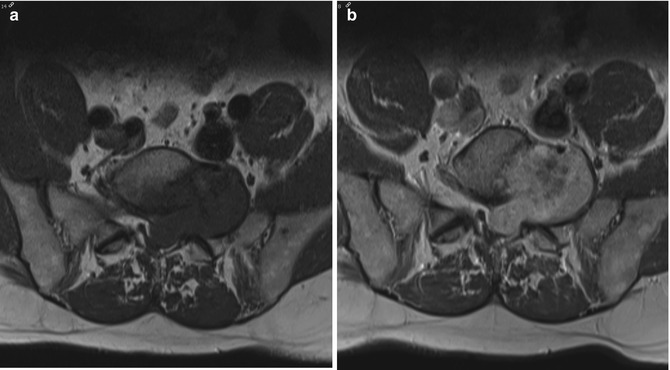
Fig. 17.2
(a, b) Schwannoma of the right hand in a 73-year-old man: (a) plain radiograph of the right hand showing a soft tissue mass between the first and second metacarpal. (b) Axial T1-weighted image (T1-WI) after gadolinium contrast injection shows a well-circumscribed soft tissue tumor with subtle peripheral enhancement
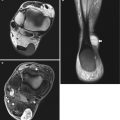
Fig. 17.3
(a–c) Schwannoma of the chest wall in a 16-year-old boy: (a) plain radiograph of the rib cage shows irregular scalloping (notching) of the inferior border of the left tenth rib and widening of the intercostal space. (b) CT scan after iodinated contrast injection shows a large, irregularly enhancing tumor with necrotic or cystic non-enhancing cavities. (c) Coronal T1-WI after gadolinium contrast injection shows a markedly enhancing tumor subjacent to the tenth rib
Fig. 17.4
(a, b) Schwannoma of the left intervertebral foramen L5–S1 in a 53-year-old male: (a) axial T1-WI shows a dumbbell-shaped lesion which causes erosion of the body of L5 and the left facet joint, resulting in widening of the left intervertebral foramen. (b) On the axial T1-WI after gadolinium contrast injection, the schwannoma shows diffuse enhancement
Ultrasound
On ultrasound, schwannomas are usually seen as solid, ovoid, hypoechoic masses with posterior acoustic enhancement. They are often surrounded by a hyperechoic capsule. Schwannomas are oriented along the long axis of the nerve with proximal and distal taillike formations (Fig. 17.5) They are usually eccentrically located in relation to the nerve axis and do not infiltrate adjacent tissue [7, 36, 73, 83, 92]. Ancient schwannomas show marked degenerative changes, such as internal bleeding, fibrosis, calcification, and cystic necrotic alterations (Fig. 17.6) [69]. In large lesions, impressive displacement of nerve fascicles and increased internal vascularization can be seen, making it sometimes difficult to rule out malignancy.
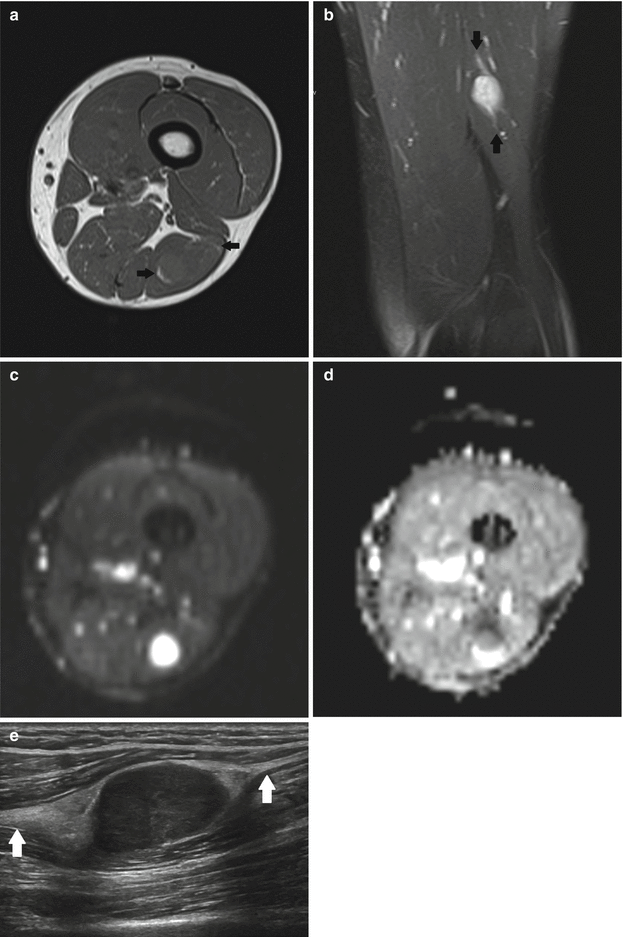
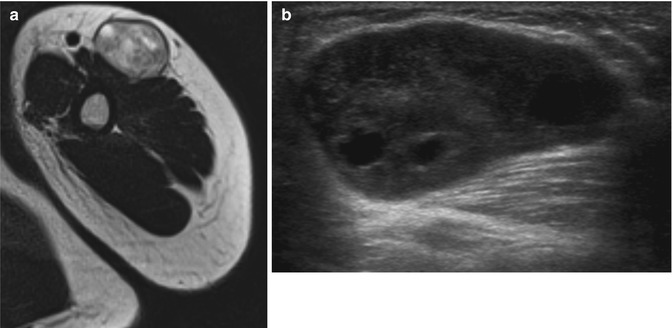
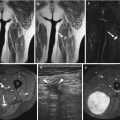
Fig. 17.5
(a–e) Cellular schwannoma of the left upper leg in a 52-year-old male: (a) axial T1-WI shows an isointense mass in the biceps femoris muscle which is partially surrounded by a thin fat rim (split-fat sign, arrows). (b) On coronal FS T2-WI, the lesion is hyperintense, displaying a fusiform shape with an entering and exiting nerve (tail sign, arrows). (c) Axial diffusion-weighted image (DWI) shows high signal intensity, which is indicative of restricted diffusion. (d) On the axial apparent diffusion coefficient (ADC) map, the lesion displays low signal intensity, confirming diffusion restriction. Although diffusion restriction may suggest a malignant peripheral nerve sheath tumors (MPNSTs), it can also be seen in benign cellular schwannomas. (e) Longitudinal ultrasound shows a hypoechoic well-circumscribed mass and also illustrates the tail sign (white arrows indicating entering and exiting nerve) and hyperechogenic rim of fat separating the tumor from the surrounding muscle
Fig. 17.6
(a, b) Ancient schwannoma of the left upper arm in a 24-year-old male: (a) axial T2-WI shows a heterogeneous mass of intermediate signal intensity and some well-circumscribed zones of high signal intensity. (b) Ultrasound shows a hypoechoic mass with intralesional nodular cystic components
Computed Tomography
On unenhanced CT scans, schwannomas are seen as well-circumscribed homogeneous lesions. They are hypo- to isodense relative to muscle. However, an inhomogeneous tumor appearance with low-density areas is frequently seen in larger tumors, reflecting the histologic diversity [19, 49].
On contrast-enhanced CT scans, most schwannomas become iso- or hyperdense to muscle (Fig. 17.7). Large schwannomas typically contain non-enhancing cystic or necrotic areas [19, 49].
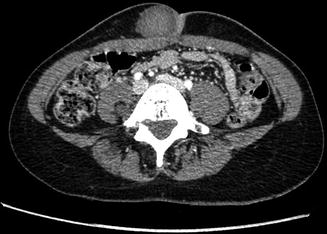
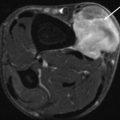
Fig. 17.7
Nerve sheath tumor in the abdominal wall in a 31-year-old female with neurofibromatosis type 2 (NF2): axial contrast-enhanced CT-image shows a well-circumscribed lesion which enhances slightly heterogeneous, displaying a density similar to muscle
Magnetic Resonance Imaging
Most peripheral nerve sheath tumors (PNSTs) share some common MR imaging characteristics (Table 17.2). However, none of these characteristics are pathognomonic for PNSTs [82].
Table 17.2
Imaging findings suggestive of neurogenic tumors
Location in the region of a nerve |
Fusiform shape |
Tail sign: entering and/or exiting nerve |
Split-fat sign (T1-weighted images) |
Target sign (T2-weighted images) |
Fascicular sign (T2-weighted images) |
Muscle atrophy/fatty replacement/edema |
On T1-weighted images (T1-WI), most PNSTs, including schwannomas, display slightly higher signal intensity than muscle, which makes them sometimes difficult to detect. On proton density-WI, they are hyperintense to muscle. On T2-WI, signal intensity is markedly increased, resulting in a sharp contrast between the tumor and adjacent fat and muscle (Fig. 17.8) [1, 3, 15, 42, 57, 71, 84, 100].
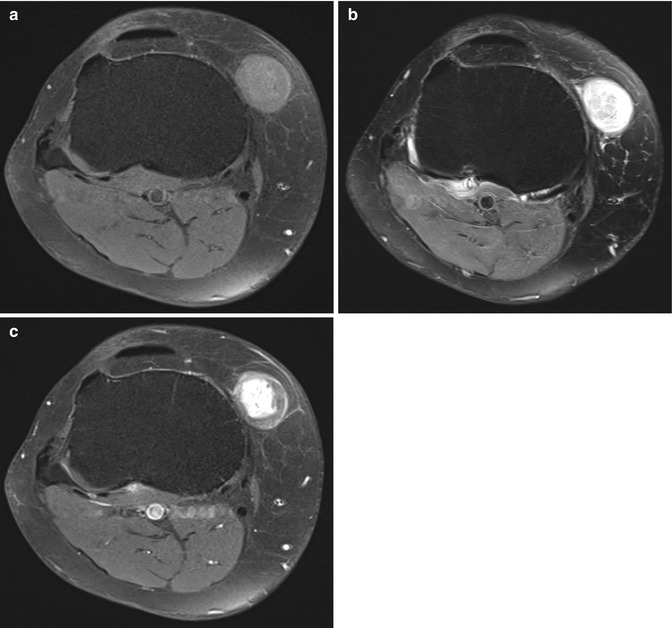
Fig. 17.8
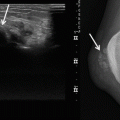
(a–c) Schwannoma of the anteromedial side of the right knee in a 58-year-old male: (a) axial FS T1-WI shows that the lesion is slightly more intense than subcutaneous fat. (b) On axial FS T2-WI, a target appearance with central hypointensity and a peripheral high signal intensity rim can be seen (target sign). (c) Axial FS T1-WI after gadolinium contrast shows central enhancement. Although the target sign and central enhancement are more commonly seen in neurofibromas, there is a lot of overlap and schwannomas may have the same signal characteristics and contrast enhancement pattern
Most nerve sheath tumors are located in the proximity of a major nerve. Due to growth along the course of the affected nerve, nerve sheath tumors typically have a fusiform shape. Oriented along the long axis of the nerve sheath tumor, the involved nerve can be seen entering and/or exiting the neoplasm, resembling a tail coming off the tumor, hence the name of this imaging feature: the tail sign. This sign has proven to be the most important imaging feature that should always suggest the diagnosis of neurogenic neoplasm. The tail sign is usually easy to detect in lesions affecting large, deep nerves, but is often difficult or impossible to assess in superficial or in small lesions (Figs. 17.9, 17.10, 17.11, and 17.12) [1, 47, 57, 65, 71, 84].
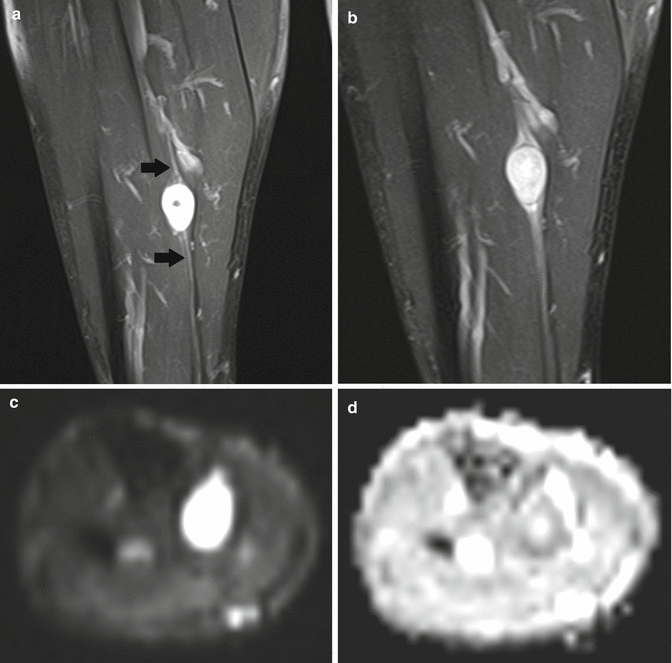
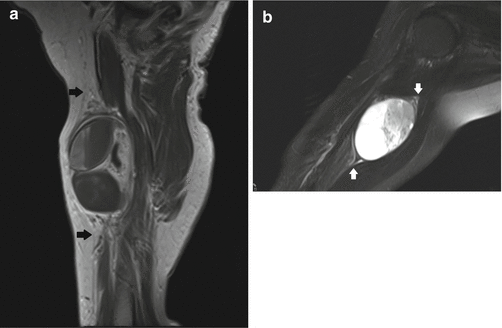
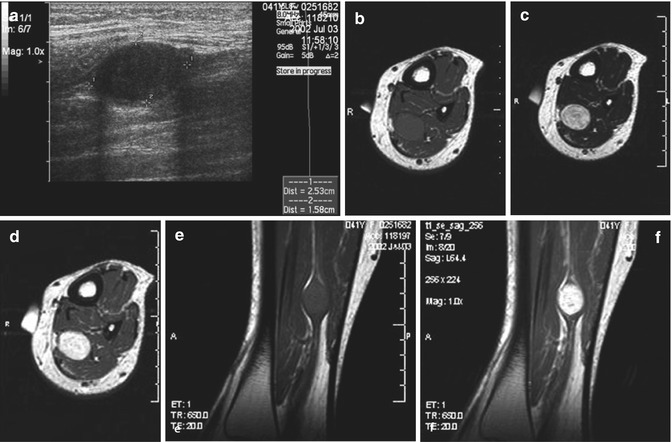
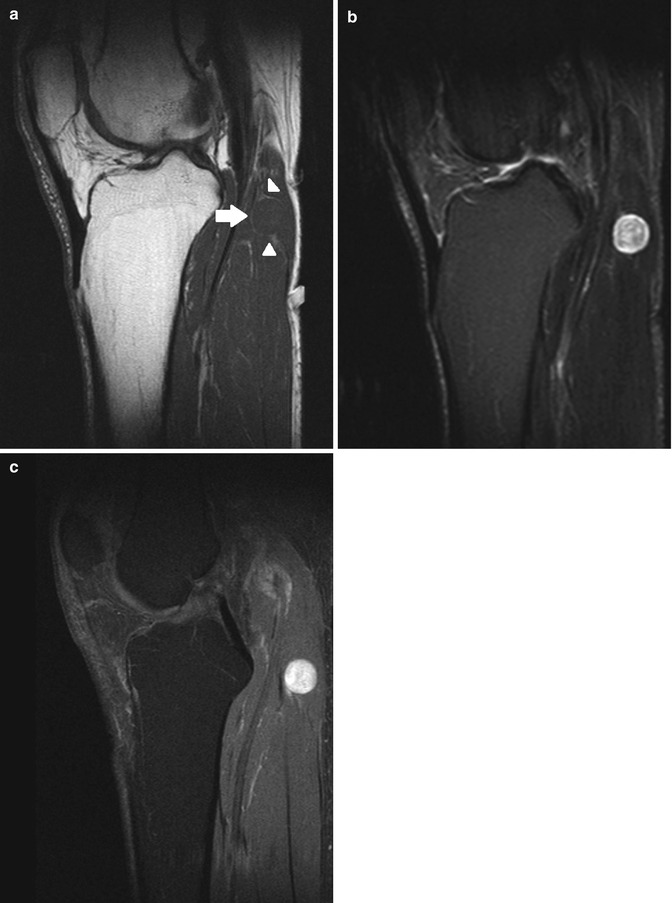
Fig. 17.9
(a–d) Ancient schwannoma of the right calf in a 60-year-old male: (a) coronal FS T2-WI shows the fusiform-shaped lesion to be located along the tibial nerve. An entering and exiting nerve can be seen (tail sign, arrows). These features are suggestive of a neural origin. (b) On coronal FS T1-WI, the lesion shows diffuse contrast enhancement. Only a small central zone shows no enhancement, compatible with central cyst formation or necrosis. (c) On the axial DWI, the schwannoma shows restricted diffusion. (d) On the axial ADC map, the lesion shows a low signal peripherally and a high signal centrally, in keeping with central cyst formation, which is often seen in long-standing (ancient) schwannomas
Fig. 17.10
(a, b) Ancient schwannoma of the right upper arm in a 76-year-old female: (a) sagittal T1-WI shows a heterogeneous, fusiform-shaped lesion. An entering and exiting nerve can be seen (tail sign, arrows). The lesion is separated from the triceps muscle by a layer of fat (split-fat sign). (b) Coronal FS T2-WI shows internal cyst formation. Again, the fusiform shape and the tail sign (arrows) can be seen
Fig. 17.11
(a–e) Schwannoma at the flexor surface of the left lower leg in a 41-year-old woman: (a) US shows a sharply defined, homogeneously hyporeflective tumor with a fusiform shape and an entering and exiting nerve (tail sign). (b) Axial T1-WI shows a well-delineated, homogeneous mass, which is isointense to the adjacent muscles. (c) On axial T2-WI, the tumor has a high signal intensity. Individual nerve fascicles can be seen (fascicular sign). (d) Axial T1-WI after gadolinium contrast injection shows homogeneous enhancement. (e) Coronal T1-WI shows a fusiform mass displaying the tail sign and split-fat sign (f) Coronal T1-WI after gadolinium contrast injection shows homogeneous enhancement
Fig. 17.12
(a–c) Neurofibroma of the left knee in a 68-year-old male: (a) on sagittal T1-WI, the lesion is isointense to the surrounding gastrocnemius muscle. Note that the lesion is partially surrounded by a small rim of fat, known as the split-fat sign (arrow). The tail sign can be seen as well (arrowheads). (b) On sagittal FS T2-WI, the lesion displays a target sign, with high signal intensity peripherally and intermediate signal intensity peripherally. (c) Sagittal FS T1-WI after gadolinium contrast enhancement shows diffuse intermediate enhancement with several irregular areas of marked enhancement
Since each neurovascular bundle is normally surrounded by fat, benign masses originating in the intermuscular space around the neurovascular bundle usually maintain a rim of fat about them as they slowly enlarge and remodel the surrounding fat plane. This rim of fat separates the tumor from the surrounding muscle tissue and appears more prominent at the tapering margins of the neoplasm. Known as the split-fat sign, this configuration suggests a tumoral origin in the intermuscular space about the neurovascular bundle, among which PNSTs are most frequent [54]. The split-fat sign is best appreciated on T1-WI as a hyperintense rim of fat. It is most seen in benign PNSTs and lesions of large nerves (Figs. 17.11 and 17.12). In malignant PNSTs, the rim of fat is typically incomplete, reflecting their infiltrative growth pattern [1, 54, 55, 57, 65, 67, 71, 103].
The target sign consists of low-to-intermediate signal intensity centrally with a ring of high signal intensity peripherally on T2-WI. Pathologically, this corresponds to fibrous tissue centrally and more myxoid tissue peripherally. Rarely, a reverse pattern of target appearance on T1-WI is seen, with central hyperintensity and peripheral hypointensity. Cutaneous PNSTs are less likely than deeper lesions to demonstrate the target sign. Initially considered pathognomonic for neurofibromas, the target sign has also been observed in schwannomas, and, rarely, malignant PNSTs (Fig. 17.8) [42, 47, 55, 57, 65, 71, 92].
The fascicular sign represents multiple small ringlike structures with low signal intensity on T2-WI, corresponding to the fascicular bundles seen in neurogenic neoplasms. The fascicular sign is best appreciated if the tumor is surrounded by fat. It is slightly more common in schwannomas than in neurofibromas, while it is not seen in malignant PNSTs (Fig. 17.11) [3, 42, 54, 55, 57].
Nerve sheath tumors may induce muscle denervation changes, including increased fat content or decreased muscle size. These changes may be quite subtle and may require comparison with the normal contralateral side. Muscle denervation changes are best seen on T1-WI. Other abnormalities of the supplied muscles, including edematous appearance, may also be seen on T2-WI [1, 84].
The following paragraph and Table 17.4 highlight the most useful MR imaging features in the differentiation between neurofibromas and schwannomas (Table 17.4).
The fascicular sign is more frequently seen in schwannomas, while the target sign is more suggestive of neurofibromas. Schwannomas tend to eccentrically displace the affected nerve, while in neurofibromas, the tumor is intimately intermixed with the nerve fascicles [36, 57, 65, 92]. However, in many situations, this distinction is not possible because both lesions often have a deep location within the epineurium and similar imaging characteristics relative to the parent nerve [42]. This parameter seems to be more reliable for discrimination between lesions in larger peripheral nerves. For all practical purposes, however, visualization of a nerve eccentrically entering a mass must be considered strongly suggestive of a schwannoma [92].
Schwannomas are more frequently encapsulated by epineurium than neurofibromas and malignant PNSTs. Encapsulation is visualized on MRI as a low signal intensity rim surrounding the tumor on T1- and T2-weighed images [65, 71]. In large schwannomas however, this finding may be difficult to appreciate since large tumors may make the capsule very thin [103].
Schwannomas have been reported to be more frequently associated with diffuse enhancement after gadolinium contrast administration, while neurofibromas might be more commonly associated with central enhancement after contrast (Fig. 17.4) [42]. However, this parameter must be evaluated with caution because of some inconstancy and contradictory results related by different reports [14, 24, 42, 55, 67]. Indeed, a recent paper reported no significant difference with respect to vascularization pattern (Figs. 17.8 and 17.9) [92].
Ancient schwannomas exhibit degenerative changes, including internal bleeding, fibrosis, calcification, and cystic necrotic alterations, resulting in a heterogeneous appearance on MR imaging (Figs. 17.6 and 17.9). This is far less seen in neurofibromas, which most commonly display homogeneous signal intensity [1].
Special histologic schwannoma types can exhibit unique MR imaging characteristics. Cystic schwannomas display low signal intensity on T1-WI and high signal intensity on proton density- and T2-WI. Solid, highly cellular tumors have intermediate signal intensity on T1-WI. Melanotic schwannomas show high signal intensity on T1-WI, reflecting T1-shortening due to the presence of melanin [44, 54, 57, 71].
17.2.2 Neurofibromas
17.2.2.1 Epidemiology
Neurofibromas represent approximately 5 % of benign soft tissue neoplasms [47]. There is no obvious predilection for any racial or ethnic group to develop neurofibromas, and these lesions have been described throughout the world [38].
Three types of neurofibromas are classically described: localized, diffuse, and plexiform. Localized neurofibromas are the most common type, representing approximately 90 % of these lesions. The vast majority of localized neurofibromas are solitary and not associated with NF1 [24]. They develop mostly between the ages of 20 and 30. Like schwannomas, they affect both sexes equally [62]. Diffuse neurofibromas are an uncommon but distinctive type that primarily affects children and young adults. The majority of diffuse neurofibromas are isolated lesions not associated with NF1. Plexiform neurofibromas are essentially pathognomonic of NF1. Development of these lesions usually occurs in early childhood. Plexiform neurofibromas have a potential for malignant degeneration and are recognized as precursors for malignant PNSTs in NF1 patients [3].
17.2.2.2 Topography
Most localized, solitary neurofibromas are superficial lesions of cutis or subcutis (Fig. 17.7) [62]. Neurofibromas in NF1 are relatively frequent on the trunk, which contrasts with the distribution of peripheral nerve schwannomas, which are more frequent in the extremities [76].
Diffuse neurofibromas demonstrate a plaque-like elevation of the skin with thickening of the entire subcutis.
Plexiform neurofibromas may involve any or all of the following sites: the cranial and spinal nerve roots and ganglia; the major nerves of the neck, trunk, and limbs, including the sympathetic system and its ganglia; and the subcutaneous branches of the major nerves and the visceral sympathetic plexuses [76]. Below the diaphragm, plexiform neurofibromas are typically located in the retroperitoneal space and paraspinal region. Typical retroperitoneal localizations are (bilateral) parapsoatic and presacral neurofibromas. Less commonly, retroperitoneal neurofibromas are found near the celiac axis and near the origin of the superior mesenteric artery [6]. Involvement of the myenteric plexuses and mesentery of the gastrointestinal tract by plexiform neurofibromas is rare. Neurofibromas in this location cause pain, intestinal bleeding, and obstruction. The regional nerves are enlarged, and nodules of varying size are found, especially in Auerbach’s plexus. The most common site is the jejunum, followed by the stomach [35].
17.2.2.3 Histology
Typically, localized neurofibromas (Table 17.3) are benign, slowly growing, and variably encapsulated neoplasms [14, 38, 62]. Neurofibromas are intimately associated with the parent nerve, growing in a longitudinal fusiform manner with the nerve entering and exiting from the lesion. Surgical resection requires sacrificing the parent nerve because the neurofibroma cannot be separated from the nerve fibers [22].
Table 17.3
Comparative pathology of schwannoma and neurofibroma
Schwannoma | Neurofibroma | |
|---|---|---|
Cell type | Schwann cells | Schwann cells and fibroblasts |
Capsule | Usually encapsulated | Usually nonencapsulated |
Extension along nerve bundle | Focal | Infiltrative growth |
Tumor shape | Round or fusiform | Fusiform |
Tumor topography | Eccentric tumor | Central tumor |
Displacing tissue along nerve axis | Separates tissue | |
Intratumoral cysts | Common | Rare |
Intratumoral necrosis | Common | Rare |
Intratumoral hemorrhage | Common | Rare |
Malignant degeneration | Very rare | More frequent |
Blood supply | Thickened arteries, prominent veins | No thickened arteries, no prominent veins |
As opposed to schwannomas, most neurofibromas are solid tumors macroscopically: areas of cystic degeneration, hypocellularity, and xanthomatous material are uncommon [76]. Neural axons traversing the tumor often follow a tortuous course. As in schwannomas, the capacity of neurofibromas for melanogenesis, although rare, has been well documented [38].
Plexiform neurofibroma relates to a diffuse enlargement and distortion of a major nerve trunk. Disorganized growth of nerves is noticed within and occasionally also outside the nerve trunk [62]. Multiple masses are found along the course of nerve bundles or trunks, with irregular cylindrical enlargement of the affected nerves [86]. Fusiform or spherical neurofibromas along their course can produce a moniliform effect or sometimes give rise to massive growths of a similar character [76].
In many neurofibromas, the peripheral zone contains rather myxoid tissue with high water content, while the central area contains more dense collagenous tissue [86]. This concept is important to understand because it is presumed to be the histologic correlate of the so-called target appearance of neurofibromas on US and MR imaging studies.
17.2.2.4 Clinical Presentation
Solitary neurofibromas present as relatively non-tender masses in the skin or subcutaneous tissue. They are somewhat mobile and moderately firm but compressible. Isolated neurofibromas in the skin usually do not exhibit an associated local change in pigmentation of the skin, although hyper- or hypopigmentation is possible. If solitary neurofibromas occur along the course of a peripheral nerve, the clinical symptoms are usually related to a space-occupying lesion but can be similar to those described for schwannoma [38]. The clinical presentation of NF-associated neurofibroma will be discussed below (see Sect. 17.4).
17.2.2.5 Imaging Characteristics
Plain Radiography
The appearance of neurofibromas on plain radiography is similar to that of schwannomas.
Ultrasound
In contrast to schwannomas, localized neurofibromas are homogeneous concentric lesions that do not displace the fascicular elements of the according nerve but rather interfere with them. On transverse images, sometimes the target sign can be seen as a hyperechoic center surrounded by a hypoechoic rim. This correlates with the ultrastructure of neurofibromas with a fibrocollagenous center surrounded by a myxomatous periphery (Fig. 17.13). Large lesions often protrude beyond the surface of the nerve, without a clear capsule [36, 65, 73].
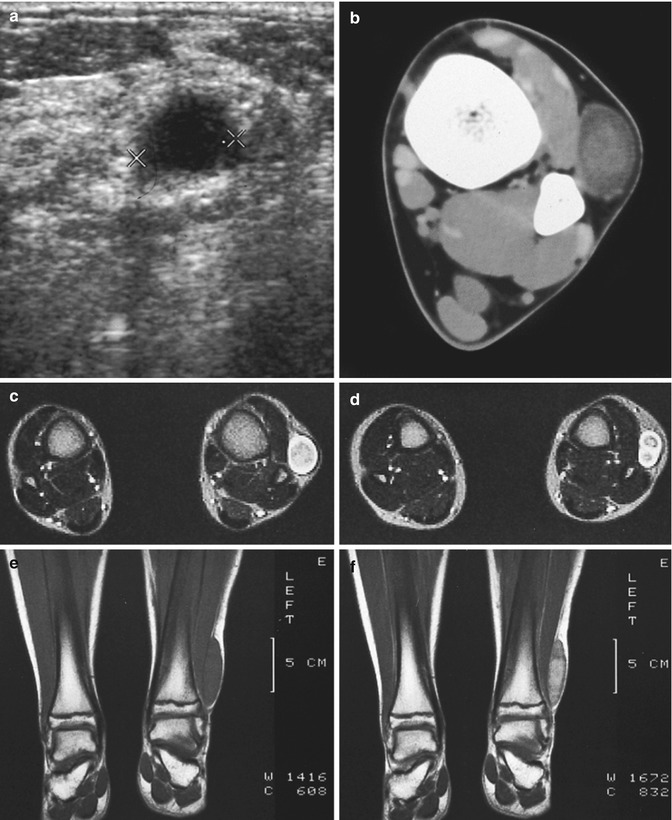
Fig. 17.13
(a–f) Neurofibroma of the cutaneous tributary of the left superficial fibular nerve in a 14-year-old boy: (a) axial ultrasound image 8 cm proximal to the left fibular malleolus shows a well-circumscribed anechoic mass. (b) CT scan after iodinated contrast injection, proximal to the left ankle joint, confirmed the mass lesion in the subcutaneous fat next to the extensor digitorum longus muscle. The central part of the lesion shows a slightly higher density than the peripheral rim (target appearance). (c) T2-WI 7 cm proximal to the ankle joints confirms the target appearance, with a central area of moderately high signal intensity surrounded by a rim of extremely high signal intensity. (d) On axial T2-WI 3 cm proximal to the ankle joints, there is bifurcation in the inferior part of the lesion. (e) Coronal T1-WI shows the tumor to be isointense relative to muscle. (f) On coronal T1-WI after gadolinium contrast injection, there is central tumor enhancement
Computed Tomography
On unenhanced CT scans, neurofibromas mostly appear as hypodense lesions due to the presence of Schwann cells, neural elements, and adipocytes [57]. Occasionally, hyperdense areas can be seen, which are believed to result from dense bands of collagen tissue produced by fibroblasts [49]. On enhanced CT scans, neurofibromas usually show little or no contrast uptake (Fig. 17.7).
Magnetic Resonance Imaging
Most neurofibromas share several imaging features with other PNSTs (Table 20.1) [1, 3, 42, 57, 71, 84, 100].
MRI is useful for the differentiation of neurofibromas and schwannomas. However, no single MR imaging finding or combination of findings allows a definitive differentiation between schwannomas and neurofibroma (Figs. 17.12 and 17.13) (Table 17.4) [1].
Table 17.4
Imaging characteristics of peripheral nerve sheath tumors (PNSTs)
Schwannoma | Neurofibroma | MPNST | |
|---|---|---|---|
CT characteristics
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |||