FIGURE 15.1-1: Embryology of primary neurulation in which neural plate develops (A) and then infolds to become a groove (B) and with closure a neural tube (C). D: Diagram of the dorsal aspect of the developing craniospinal area in the embryo.
ACRANIA/EXENCEPHALY-ANENCEPHALY
Description: Acrania is a defect that occurs due to complete or partial absence in the development of the cranial vault above the orbits, with absence of parietal, squamousal, occipital, temporal, and frontal bones above the supraciliary ridge.9 Exencephaly is acrania with protrusion of substantial brain into the amniotic cavity.10–12 Anencephaly represents absence of forebrain, midbrain, and skull. The cerebral hemispheres are replaced with residual covering of hemorrhagic, fibrotic, degenerated neurons and glia with little definable structure (Fig. 15.1-2).
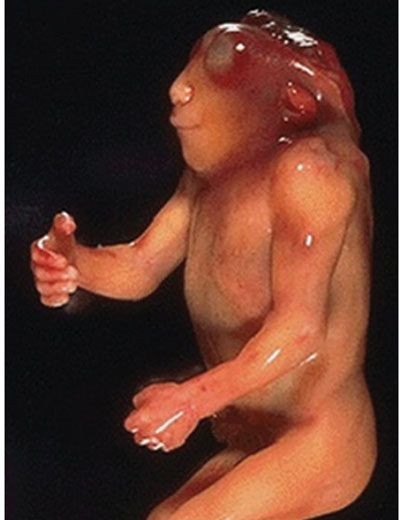
FIGURE 15.1-2: Postmortem of a fetus with anencephaly.
Incidence: The incidence of anencephaly is difficult to assess, but in the United States, frequency is estimated at 1 in 1,000 pregnancies.13 A progressive decline is suspected with periconceptional folate fortification and termination.14,15 Anencephaly has a higher rate in countries such as Egypt, Lebanon, Ireland, Scotland, and New Zealand. In the United States, fetuses of Caucasian and Hispanic ethnicity are more frequently affected than those of African descent.16 Twin gestations also have a higher risk for anencephaly. There is a female preponderance of 3:1.15,16 Ninety-five percent of infants born with anencephaly are in families with no prior history for a NTD.15
Pathogenesis: Exencephaly–anencephaly sequence is the most severe NTD and results from failed closure of the rostral end of the neural tube. The severity of the bone defect is variable and may result in absent brain or rudimentary disorganized brain. Exencephaly pathologically demonstrates a highly vascular layer of epithelium with two relatively equivalent disorganized, dysplastic cerebral hemispheres.12,13,17 With progression to anencephaly, the cerebral hemispheres are replaced by a mass of connective and vascular tissue with scattered islands of brain tissue, so-called angiomatous stroma or area cerebrovasculosa.17,18
Acrania is a developmental anomaly that is characterized by partial or complete absence of the cranium with complete but abnormal development of the cerebral tissue. This disorder is hypothesized to occur at the beginning of the 4th week at the same time the anterior neuropore closes, but is related to failure of the mesenchyme to migrate under the ectoderm and superficial to the cerebral hemispheres.8 The cerebral hemispheres are present but disorganized, and the brain is covered only by a thin membrane.
The major insult in acrania, exencephaly, and anencephy is due to lack of cranial development. The cerebral tissue is not protected by meninges, skull, and skin, and is progressively destroyed through exposure to amniotic fluid and mechanical trauma.17 As a result, the brain disappears from 14 weeks onward.19,20 Prior animal and US reports have demonstrated that there typically is progression of acrania-exencephaly to anencephaly (AEA).7,17,21 In some cases, residual disorganized brain may persist late in gestation or even postnatal.
Etiology: AEA appears to be multifactorial in origin, being from both genetic and environmental causes.9 Associated chromosomal syndromes have been reported in 5% to 10% of these cases and include trisomy, triploidy, mosaic trisomy 11 and 20, and a number of deletions and duplications or single gene disorders.7 Hyperthermia, deficiency in zinc and copper, and occupation solvent exposure have been associated with a higher risk of anencephaly.14,20 Most relevant is inadequate dietary consumption of folates prior to conception.7,13,14
Diagnosis: Preliminary screening for high maternal serum alpha-fetoprotein (AFP) at 10 to 15 weeks may disclose evidence of leakage from the fetal neural tube in 90% of cases.22 The combination of elevated AFP and low estriol levels is highly predictive for anencephaly.22,23 However, screening can be falsely positive and may require confirmation with amniocentesis or US.
Ultrasound: Sonography can identify almost 100% of anencephalic fetuses.6,9,21,24 The disorder can be diagnosed transvaginally prior to 10 weeks’ gestation by the presence of a widened cranial pole, altered brain echotexture, variable asymmetric or lobulated disorganized tissue, and decreased head-to-trunk ratio.24,25 However, confirmation is usually obtained after 11 to 12 weeks’ gestation when calvarial ossification defined as a hyperechogenic structure compared with the underlying soft tissues is noted to be absent.6,21,24,25 Detection may also improve after 14 weeks when the brain has completely formed, although in AEA a moderate to large amount of disorganized cranial tissue may be present early in gestation as it has not yet been destroyed by amniotic fluid.19 From 10 to 14 weeks, the “Mickey Mouse” sign can be seen on coronal images, as cerebral lobes floating in amniotic fluid above the orbits (Fig. 15.1-3).26 AEA may result in a reduced crown rump length or chin length-to-crown rump length ratio, but in some cases the measurement is normal and can lead to a false negative study below 14 weeks.19,26–28 Echogenic amniotic fluid, believed to represent particles of degenerated brain, has also been described in the first trimester.20
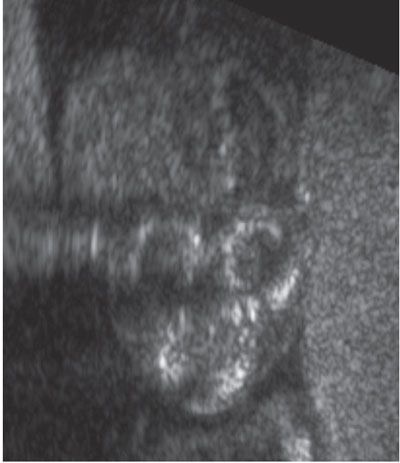
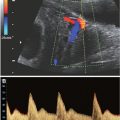
FIGURE 15.1-3: Coronal US of acrania/exencephaly with absent calvarium and protruding brain above orbits giving rise to Mickey Mouse appearance.
In the second trimester, the diagnosis is easier as less brain tissue is present. The typical “frog eyes” sign on coronal plane is due to prominent orbits in conjunction with the symmetric absence of a normally formed calvarium and brain (Fig. 15.1-4).18 Exposed residual neural tissue may be echogenic, cystic, or normally formed with pseudosulcations.12,18 The cerebellar hemispheres may be absent, and spinal segmentation anomalies are common, especially in the cervical area. In the second or third trimester, as many as 30% to 50% of cases have associated polyhydramnios from impaired fetal swallowing, excess CSF across the meninges, or increased fetal urine output due to absent antidiuretic hormone.18
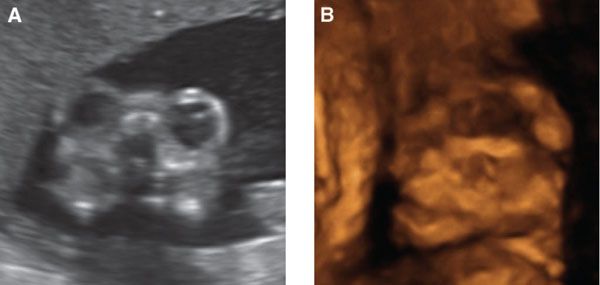
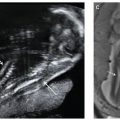
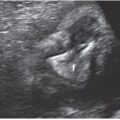
FIGURE 15.1-4: Anencephaly A: Coronal 2D image shows prominent “frog eye” appearance. B: 3D image demonstrating lack of skull above the orbits.
MRI: Fetal MRI is usually not required to confirm the diagnosis of anencephaly. MRI may be utilized when there is limitation in US imaging (obesity) or in the case of medical, legal, or ethical issues.29 It can be helpful to exclude other pathologies mimicking AEA and useful in multiple gestations when it is necessary to confirm normal development of the co-twin.
The calvarium is absent, and there may be disorganized brain tissue in the area of the cerebral hemispheres in the presence of acrania or exencephaly (Fig. 15.1-5). With anencephaly, the orbits are prominent, no brain tissue is seen, and a cervical spine defect with absence of brainstem and spinal cord can be identified (Fig. 15.1-6).
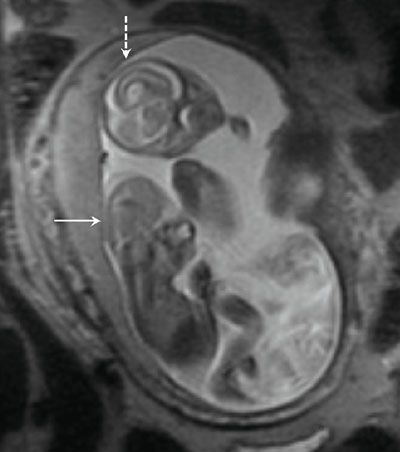
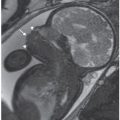
FIGURE 15.1-5: SSFSE T2 fetal MR showing lack of calvarium and dysmorphic brain in acrania/exencephaly (solid arrow) and normal brain and calvarium in co-twin (dashed arrow).
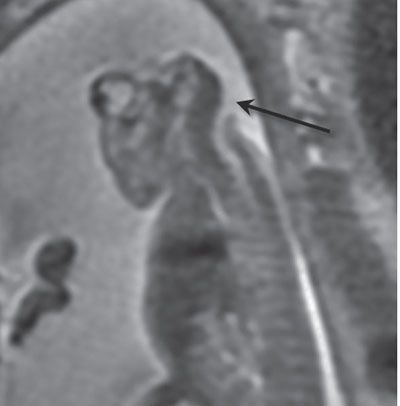
FIGURE 15.1-6: SSFSE T2 sagittal image of fetus with anencephaly demonstrating cervical spine defect and absent brainstem and cervical cord (arrow).
Associated Anomalies: Associated spinal lesions can be found in up to 50% of cases. Other common anomalies include cleft lip/palate, cardiac, gastrointestinal, clubfoot, and omphalocele.8,9,30
Differential Diagnosis: Differential includes large cephaloceles, osteogenesis imperfecta (OI), and hypophosphatasia. In both OI and hypophosphatasia, the bones are present but poorly mineralized, and other fractures, bone shortening, and/or bowing are typically present.
This disorder should be distinguished from amniotic band syndrome, in which there is an asymmetric brain defect, multiple limb or digit amputations, asymmetric ventral wall defects, and unusual craniofacial or spinal defects.11 Amniotic band is often associated with oligohydramnios, which is rare in anencephaly.18
Prognosis: Approximately 65% of pregnancies with anencephaly die in utero.20 Affected fetuses born alive have a rudimentary brainstem. The brainstem can support reflex actions such as breathing and sometimes responses to sound and touch. Children with AEA are not viable and have only short-term survival.13
Management: Elective termination is often considered. In the United States, prenatal diagnosis and pregnancy termination have decreased the prevalence of anencephaly at birth by 60% to 70%. In Europe, America, and Asia, the overall frequency for termination of anencephaly pregnancy is 83%, ranging from 59% to 100%.30,31
Labor and delivery are commonly associated with unstable fetal lie, dysfunctional labor, shoulder dystocia, and postpartum hemorrhage.32 Up to 35% of anencephalic infants will die during labor. For the 45% that are born alive, supportive care is typically provided for minutes to days.32 The potential for neonatal organ donation has raised both legal and ethical issues.
Recurrence Risk: There is a 2% to 5% recurrence risk.9,15 Increased risk also exists for those with relatives affected by anencephaly or those with genetic predisposition.
CEPHALOCELES
Description: A cephalocele is a defect in the skull and dura with extracalvarial extension of intracranial structures enclosed by overlying skin.
There are four major types.33,34 Meningoencephaloceles, the most common, are herniations of cerebrospinal fluid (CSF), brain, and meninges through a calvarial defect. If an encephalocele includes part of a ventricle and choroid plexus, it is termed meningoencephalocystocele. Meningoceles are herniations of only meninges and CSF. An atretic cephalocele is typically parieto-occipital and consists of a tract and small defect of dura, fibrous tissue, and degenerated brain. Glioceles are glial-lined CSF defects.
Incidence: Cephaloceles account for 10% to 20% of craniospinal dysraphisms. Estimated prevalence is 0.8 to 4 per 10,000 live births.39 The actual incidence may be higher as many of these malformations result in termination, in utero demise, and stillbirth.40 There is a difference in incidence geographically. The Western hemisphere reports 1 to 3 per 10,000 births, whereas Southeast Asia reports 1 in 5,000 births.28 In the West, occipital encephaloceles are most common (80%), while midline frontal and parietal are evenly divided between the remaining 20%.41 Females are affected twice as commonly as males in the occipital location.39,42 In Southeast Asia and Russia, frontal cephaloceles are most prevalent.43
Pathogenesis: Cephaloceles occur early in embryogenesis, at between 24 and 60 days.40,44 Several theories have been suggested. Many believe that a cephalocele occurs because of a failure of neural tube closure.44,45 Occipital and parietal cephaloceles, sometimes associated with other craniospinal defects, are more likely to develop from this pathophysiology.34,39 However, the presence of skin over the defect raises the question of a mesodermal insufficiency, with herniation of brain and meninges due to a primary defect in the bone and dura.35,36,43,46,47 Anterior cephaloceles may be mesodermal in origin as they are not commonly associated with other NTDs.36 A third theory emphasizes the guidance of genes, stating that variation in the pattern of cranial neural tube closure is a genetically determined factor.48
Etiology: Cephaloceles have been associated with both genetic and environmental teratogens. The defect can develop owing to exposure to trypan blue, irradiation, excess vitamin A, folic acid antagonists, triamcinolone, warfarin exposure, hyperthermia, and malnutrition.35,47 Cephaloceles are seen at a higher rate with increased maternal age, maternal diabetes mellitus, rubella, and consanguineous marriages.47 Geographic differences in the distribution of encephaloceles suggest racial and other environmental effects.39 Although most cephaloceles are sporadic, some are part of recognized genetic and nongenetic syndromes (Table 15.1-1).43,49
Syndromes/Association in Cephaloceles
Syndrome/Association | Genetic Transmission |
Occipital | |
Meckel–Gruber | Autosomal recessive |
Knobloch | Autosomal recessive |
Walker–Warburg (Chemke, HARD ± E) | Autosomal recessive |
Cryptophthalmos | Autosomal recessive |
Dyssegmental dwarfism | Autosomal recessive |
Von Voss | Autosomal recessive |
Joubert | Primary autosomal recessive |
Klippel–Feil | Variable |
Craniostenosis | Variable syndrome |
Hemifacial microsomia (oculoauriculovertebral) | Sporadic |
Ectrodactyly–ectodermal dysplasia | Sporadic and familial |
Warfarin embryopathy | |
Dandy–Walker | |
Arnold–Chiari | |
Iniencephaly | |
Myelomeningocele | |
Parietal | |
Absent corpus callosum | |
Atretic form | |