Fig. 12.1
Tijeras-Raballand—overview of the mechanisms of primary and acquired resistance to antiangiogenic agents
Primary resistance may be due to the fact that some tumors produce redundant pro-angiogenic factors besides VEGF and are thus relatively insensitive to VEGF-dependent pathway inhibition. Moreover, tumors may exhibit modes of vessel growth (such as vessel co-option, vascular mimicry, and intussusception) that are less dependent on VEGF pathway and then less sensitive to its blockade [6]. Effects of anti-VEGF agents may also be impaired by preexisting infiltration of inflammatory cells. More precisely, M2-type macrophages express various factors, such as MMP-9, CXC chemokines, and pro-inflammatory cytokines (IL-6, TNF-α, IL-1), that promote angiogenesis and may protect tumor vessels from VEGF pathway inhibition. Finally, some tumor cells can be indifferent to hypoxia. For example, pancreatic ductal adenocarcinomas primarily display a poorly vascularized stroma. Such vascular hypoxic microenvironments promote survival of anaerobic tumor cells that are intrinsically refractory to antiangiogenic treatment.
Other tumors may acquire resistance, thus become unresponsive to the antiangiogenic treatment. Most experimental evidence suggests that at least four distinct adaptive mechanisms are involved in acquired resistance to antiangiogenic agents [3]. The first mechanism is hypoxia-induced activation and/or upregulation of alternative pro-angiogenic pathways such as angiopoietin, ephrins, or FGF. Second, hypoxia also triggers recruitment of bone marrow-derived cells that can contribute to tumor vascularization in a VEGF-independent way. Third, increase in pericyte coverage of the tumor vasculature can protect endothelial cells from death consequent to the lack of VEGF-mediated survival signaling [7]. Finally, hypoxia may enhance tumor cells invasiveness into local tissue and metastatic seeding to lymph nodes and distant organs, providing access to non-tumoral tissue with mature vasculature, less sensitive to anti-VEGF therapy.
mTOR Inhibitors
The PI3K/AKT/mTOR pathway mediates signal transduction downstream many growth factor receptors. mTOR functions as a sensor of nutritional/metabolic stress during cell growth and promotes survival and protein synthesis during nutrient or energy rich periods (Fig. 12.2) [8]. This pathway is often deregulated in cancer and thus represents an interesting therapeutic target [9]. Rapalogs, which include rapamycin and associated analogs (temsirolimus, everolimus, and deferolimus), inhibit mTOR activity through association with its partner FK506 binding protein 12 (FKBP-12) [9, 10]. However, as observed with antiangiogenic agents, clinical benefit of rapalogs remains sporadic. Therefore, attempts have been made to identify predictive markers in response to rapalogs and to understand resistance mechanisms.
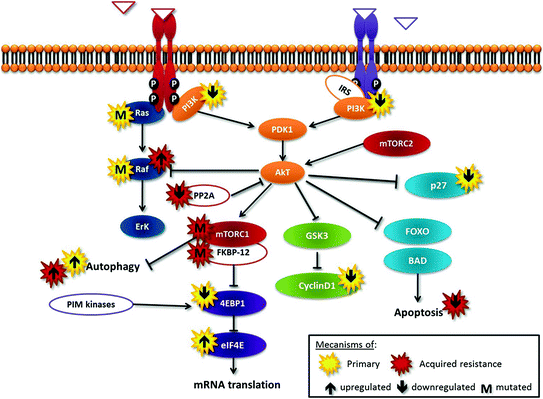
Fig. 12.2
Tijeras-Raballand—schematic representation of the Pi3K-Akt-mTOR cascade deregulations and relevant cross talks involved in resistance to mTOR inhibitors
Primary resistance may be due to activation of alternative pathways, mainly Erk pathway, such as in tumors harboring KRas or BRaf mutations. Second, one of the primary downstream substrates of mTOR, 4EBP1, suppresses eIF4E activity and then represses mRNA translation; low levels of 4EBP1 or high levels of eIF4E may confer resistance to rapalogs [8]. Third, rapamycin treatment prevents p27 downregulation, thereby inhibiting proliferation; accordingly, cells with low levels of p27 may be less responsive to rapalog-mediated growth inhibition [8]. Moreover, tumor cells in which apoptotic pathways are nonfunctional are resistant to rapalog-induced cell death. In contrast, to cells with constitutive activation of PI3K/AKT/mTOR pathway, cyclin D1 overexpression and/or functional apoptotic pathways have been shown to be more sensitive to mTOR inhibition [11].
Many mechanisms of acquired resistance to rapalog therapy have been identified that bypass, within the mTOR pathway, the blockade of the target. They include: (1) activation of feedback loops and alternative pathways (e.g., PIM kinases that can phosphorylate and activate 4EBP1 in an mTOR-independent way), (2) mutations in rapalog targets FKBP-12 or mTOR, (3) loss of function of PP2A, a phosphatase involved in dephosphorylation and inactivation of AKT, and (4) stimulation of autophagy, which is competitive to apoptosis [8].
Pathological and Molecular Characteristics of PNETs Involved in Resistance to Antiangiogenic Agents and mTOR Inhibitors
Resistance to Antiangiogenic Agents
Well-differentiated PNETs are highly vascularized tumors as measured by CD31 immunoassay and express high levels of VEGF, VEGFR-2 and VEGFR-3 [12]. They thus appear as good candidates for antiangiogenic therapy.
The transcription factor hypoxia-inducible factor-1alpha (HIF-1α) is one of the key drivers of angiogenesis in PNETs. The classical HIF targets (also known as hypoxia-related genes) include (1) genes which increase oxygen supply and delivery, such as VEGF and erythropoietin (EPO), and (2) genes that induce glycolysis, such as GLUT-1, hexokinase, and PDK-1. HIF-1α is regulated by the von Hippel–Lindau protein (pVHL), which forms the recognition component of an E3 ubiquitin ligase complex (Fig. 12.3). Under normoxic conditions, HIF-1α is hydroxylated on critical proline residues by prolyl-hydroxylase domain (PHD) proteins, which act as oxygen sensors. Following prolyl-hydroxylation, pVHL binds to HIF-1α, leading to its ubiquitination and subsequent proteasome degradation. Hypoxia-related genes are thus silenced. Conversely, under hypoxia, HIF-1α is no longer hydroxylated by PHDs and cannot be recognized by pVHL. This results in expression of hypoxia-related genes [13].
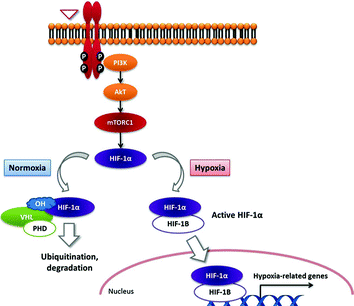
Fig. 12.3
Tijeras-Raballand—schematic representation of HIF-1 transcription factor regulation under hypoxic and normoxic conditions and the role of VHL
Two distinct mechanisms can lead to HIF-1α activation in PNETs (Fig. 12.4). First, VHL gene can be inactivated by genetic or epigenetic mechanisms, resulting in the accumulation of HIF-1α and transcription of hypoxia-related genes regardless of the oxygenation status [3, 14]. Hereditary PNETs are observed in VHL disease, a rare autosomal dominant neoplastic syndrome (about one in 36,000 live births) caused by germline mutations in the VHL gene on the short arm of chromosome 3 [15]. It is characterized by the development of various benign and malignant tumors and cysts in multiple organs: hemangioblastomas of the central nervous system, retinal angiomas, pancreatic cysts and neuroendocrine tumors, renal cell carcinoma and cysts, epididymal cystadenomas, phaeochromocytomas, and endolymphatic sac tumors [15]. PNETs occur in 10–15 % of patients with VHL disease and are frequently multiple (>30 %). VHL-associated tumors (including PNETs) are usually highly vascularized, due to upregulation of hypoxia-related genes. Alternatively, somatic mutations of VHL gene were considered to be a rare event in sporadic PNETs [8, 16]. However, Schmitt and colleagues [17] have recently reported that up to 25 % of sporadic PNETs display genomic alterations of the VHL gene that resulted in gene silencing, such as deletion and promoter methylation. These genomic abnormalities were associated with underexpression of VHL-RNA in 25 % of these tumors. Consistently, approximately one-third of tumor samples showed positive staining for HIF-1α and its targets CA9 and GLUT-1 [17].
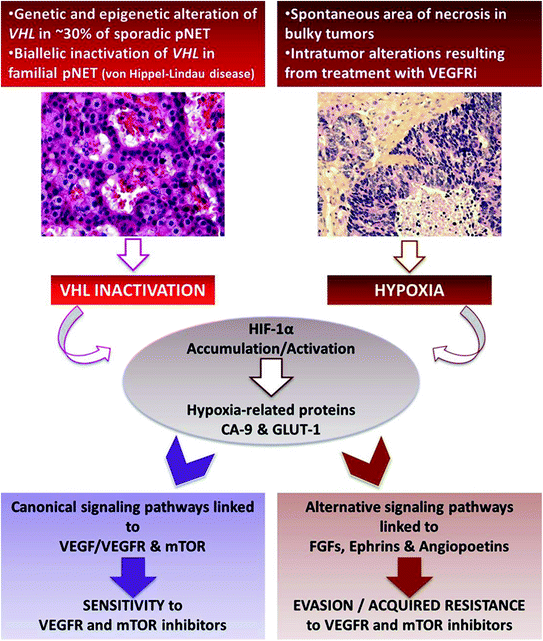
Fig. 12.4
Tijeras-Raballand—VHL inactivation and hypoxia as important events associated with pancreatic neuroendocrine tumor aggressiveness and resistance to targeted therapies
Following HIF-1α activation, canonical pathways linked to VEGF/VEGFR are activated and drive tumor angiogenesis. Thus, this uncontrolled activation of HIF-1α may convey sensitivity to anti-VEGF inhibitors. These data gave rationale for anti-VEGF-targeted therapies in PNETs and was confirmed in preclinical and clinical studies [1, 16, 18].
Alternatively, HIF-1α activation can result from tumor hypoxia. This is observed at late stage of bulky tumors’ natural history in which central hypoxia followed by necrosis spontaneously occurs, due to high tumor volume. Interestingly, high expression of PHD proteins, reflecting tumor hypoxia, has been shown to be associated with higher risks of recurrence and poor survival [13]. Hypoxia-induced activation of HIF-1α triggers canonical signaling pathways (hypoxia-related genes) but also alternative pathways, which are involved in angiogenesis, epithelial-to-mesenchymal transition (EMT), and cell survival, thus increasing tumor aggressiveness. Noticeably, hypoxia can also be the consequence of antiangiogenic treatments and thereby contribute to acquired resistance to VEGFR inhibitors. Indeed, it has been shown that several pro-angiogenic factors, including predominantly fibroblast growth factors (FGFs), ephrins, and angiopoietins (alternative-pathway genes), were upregulated in resistant tumors to VEGFR inhibitors [16]. All together, these results suggest that VEGFR inhibitor-induced hypoxia may upregulate multiple pro-angiogenic alternative factors and trigger a transition toward a resistant and more invasive phenotype.
Consequently, HIF-1α activation can be both a factor of sensitivity (when hypoxia-unrelated) and resistance (when resulting from tumor hypoxia) to anti-VEGFR therapy in PNETs.
Resistance to mTOR Inhibitors
The same ambivalence regarding consequences of HIF-1α activation exists in response to mTOR inhibitors. Experimentally, mTOR inhibition decreases HIF-1α levels. Thus, tumors that express high level of HIF-1α, such as tumors harboring VHL inactivation, may be hypersensitive to rapalog therapy. Alternatively, one of the major mechanisms proposed to underlie the anticancer activity of rapalogs is an antiangiogenic effect by inhibiting signal transduction downstream VEGFR and PDGFR. This latter leads to hypoxia, induction of HIF-1α, and its “collateral damages” as described above and may drive tumor resistance toward these agents.
Development of a Preclinical Model for PNETs Study
Preclinical models available for PNETs study are quite limited as compared with other tumor types. Indeed, PNET cell lines are difficult to obtain. No human cell line has been established to date, and only a few murine, hamster, or rat cell lines have been developed. Most murine cell lines required an SV40-based transformation. Regarding in vivo studies, almost all of them have been performed using the specific RIP1-Tag2 (RT2) mouse model developed by Hanahan et al. [19]. They used a fusion transgene composed of the SV40 large-T oncogene and the insulin-promoter, yielding to transgenic mice. Following oncoprotein expression, these mice developed multifocal islet cell dysplasia and in situ carcinomas, ultimately resulting in invasive, yet potentially lethal, PNETs. These tumors evolved according to a multistage carcinogenesis mimicking human PNETs [19]. Noticeably, as observed in human PNETs, this model reproduced the crucial step of angiogenic switch.
This transgenic model offers the opportunity to explore murine tumor cells within their physiological tissue environment. Moreover, this model appears particularly interesting to study the interactions between cancer cells and “non-tumor” cells, including those responsible for tumor angiogenesis. The RT2 model also appears highly reproducible and, therefore, represents a dedicated tool to explore pharmacological strategies targeting different stages of endocrine pancreatic carcinogenesis [16, 20–22].
A possible limitation of the RT2 model is that unlike human bulky PNETs, tumor necrosis is infrequent, leading to limited hypoxia-related signaling activation. Another limitation is the relatively low incidence of distant metastases at late stage, as mice mainly die from tumor-induced hypoglycemia. Therefore, an interesting variant of the RT2 model was developed and consisted of a double transgenic RIP1-Tag2 and RIP7-Igf-1R mouse model, overexpressing the type I insulin-like growth factor receptor (IGF-1R) in pancreatic islets [23]. This particular phenotype yields to more invasive and metastatic tumor patterns. Although the life span of these latter mice is slightly reduced in comparison with their RT2 counterparts, this model may be interesting to consider since it reproduces features close to the latest stages of human PNETs.
RT2 model has been widely used to study multiple signaling pathways—mainly related to IGF-II/IGF-1R, MMP-9 and MMP-2, VEGF-A/VEGFR2, mTOR, EGFR, and possibly PDGF-B/PDGFRβ [23–26]—and several cell types—tumor and stromal cells, endothelial cells, and pericytes [21, 24, 26]—involved in PNET development. These findings reinforce the rationale for development of targeted therapies directed against VEGF and mTOR pathways.
Another in vivo model has been marginally used. Rat pancreas was treated with azaserine, leading to a chimio-induced carcinoma. CA20948 rat PNET cell line was, thereby, established by isolating cells of acinar origin [27]. This cell line was then injected subcutaneously in the lower flank of rats. This heterotopic model does not allow the study of tumor/stroma interactions, which represents its major limitation for drug evaluation.
VEGF Pathway Inhibitors
In PNETs, antiangiogenic agents have been exclusively studied in the RT2 model. VEGF pathway inhibition was shown to decrease angiogenesis and tumor development.
In prevention experiments, VEGFR2 inhibition by anti-VEGFR2 antibody almost completely abrogated the angiogenic switch and reduced by more than 50 % the tumor burden in both intervention and regression experiments. These effects were associated with a significant reduction in vessel density and permeability [16].
Related posts:
 New Anticancer Agents in Neuroendocrine Tumors
Inhibition of mTOR in Neuroendocrine Neoplasms of the Digestive Tract
Clinical Management of Targeted Therapies in Neuroendocrine Tumours
New Anticancer Agents in Neuroendocrine Tumors
Inhibition of mTOR in Neuroendocrine Neoplasms of the Digestive Tract
Clinical Management of Targeted Therapies in Neuroendocrine Tumours
 Liver-Directed Therapies in Neuroendocrine Tumors
Liver-Directed Therapies in Neuroendocrine Tumors
 Measuring the Relationship of Quality of Life and Health Status: Including Tumor Burden, Symptoms, and Biochemical Measures in Patients with Neuroendocrine Tumors
Measuring the Relationship of Quality of Life and Health Status: Including Tumor Burden, Symptoms, and Biochemical Measures in Patients with Neuroendocrine Tumors
 Imaging of Neuroendocrine Tumors and Challenges in Response Evaluation for Targeted Therapies
Imaging of Neuroendocrine Tumors and Challenges in Response Evaluation for Targeted Therapies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


