CHAPTER 1 Pathogenesis of vascular diseases
Arteries
Normal structure
Human arteries are composed of three layers (Fig. 1-1). The intima consists of a sheet of endothelial cells lining the vessel lumen and a thin subendothelial matrix. The endothelium has a variety of critical functions.1 It controls hemostasis largely by acting as a barrier between circulating blood and the thrombogenic subendothelial layer. Endothelial cells can indirectly alter vessel caliber when changes in blood oxygen tension, pressure, or flow are detected. The endothelium produces and responds to a variety of factors that are vital to arterial repair after injury.

The media is separated from the intima by the internal elastic lamina. This layer is primarily composed of collagen, elastin, and smooth muscle cells arranged in longitudinal and circumferential bundles. Elastic (conduit) arteries (i.e., aorta, aortic arch vessels, iliac artery, and pulmonary arteries) propel blood forward because dense bands of elastin let these vessels expand during systole and contract during diastole.2 In the smaller-caliber muscular arteries, smooth muscle cells predominate, and the circumferential orientation of the cells allows the lumen to dilate or constrict in response to various stimuli.
Small arteries become arterioles, which are 40 to 200 micrometers (μm) in diameter. Arterioles lead into capillaries. Direct arteriovenous communications without interposed capillary networks exist in some vascular beds. These connections allow diversion of blood away from certain parts of the body in physiologic and pathologic states, such as shunting of blood from the skin and extremities in a hypotensive individual.
Functional disorders
Arterial tone and luminal diameter are regulated by several mechanisms1:
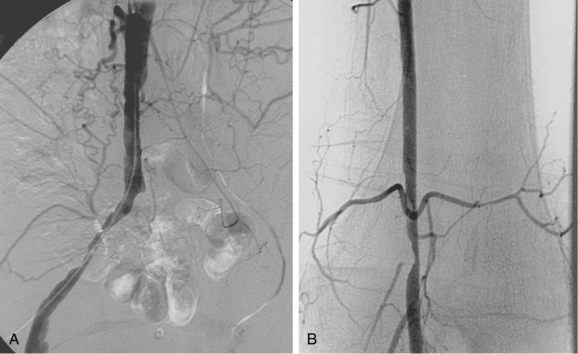
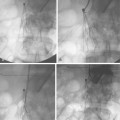
Vasodilation is seen primarily in low-resistance systems, such as arteriovenous fistulas, arteriovenous malformations, and hypervascular tumors, and in collateral circulations (Fig. 1-2). Vasoconstriction usually is the result of vascular trauma or low-flow conditions (Fig. 1-3). Ingestion or infusion of certain drugs (e.g., vasopressin, dopamine, epinephrine) also can lead to vasospasm (Fig. 1-4). Raynaud disease is a functional disorder primarily affecting the small arteries of the hands and feet in which intermittent vasospasm is caused by external stimuli3 (see Chapter 9). The hallmarks of vasospasm are resolution over time or relief with vasodilators.
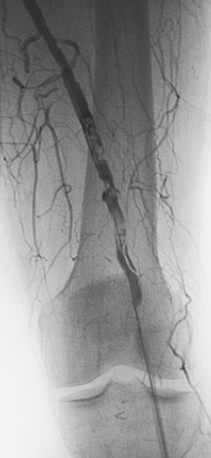
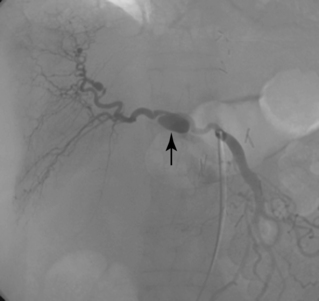
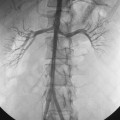
Arterial “standing” or “stationary” waves are an imaging curiosity that may be confused with functional vasospasm4 (Fig. 1-5). Standing waves have been noted at both catheter and magnetic resonance (MR) angiography. Their precise cause is unknown, but a leading hypothesis invokes secondary retrograde flow (typical in high resistance arteries) as the explanation for this temporary oscillating pattern.5,6
Atherosclerosis
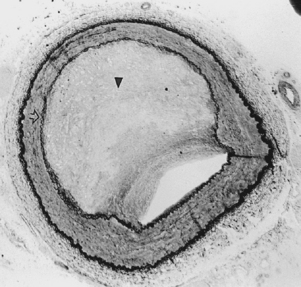
Atherosclerosis is the most common disease affecting the vascular system and the leading cause of morbidity and mortality in the Western world. It develops as a result of an inflammatory response to lipid storage in the arterial wall.7 Various provocative factors for disease may act as triggers for inflammation (see later discussion). The common inciting event is endothelial dysfunction mediated through the immune system. The damaged endothelium induces platelet activation, which in turn causes white blood cell adhesion to the vascular wall.8 Lipoproteins and monocytes enter the subendothelial space and produce “fatty streaks” composed largely of foam cells.9 A variety of factors are released in response to this pathologic process. These substances cause medial smooth muscle cells to migrate to and then proliferate in the intima. Overproduction of collagen, elastin, and proteoglycans gives the lesion a fibrotic character. Chronic inflammation ensues. With time, medial thinning, cellular necrosis, and plaque calcification and degeneration occur (Fig. 1-6). Ultimately, plaque fracture, ulceration, hemorrhage, or thrombosis may occur.
A number of risk factors for atherosclerosis have been identified (Box 1-1). However, these conditions do not account for all cases of the disease. There are several other markers for the development of atherosclerosis.10C-reactive protein (CRP) is an acute phase reactant that accumulates in blood in the presence of an inflammatory state. Elevated serum CRP is strongly associated with progression of atherosclerotic lesions in asymptomatic patients and in those with established disease.11 High levels of the amino acid homocysteine are also associated with a significant risk of arterial and venous thrombosis. Finally, increased arterial stiffness (i.e., diminished arterial distensibility) is now recognized as an important independent predictor for future cardiovascular disease.12
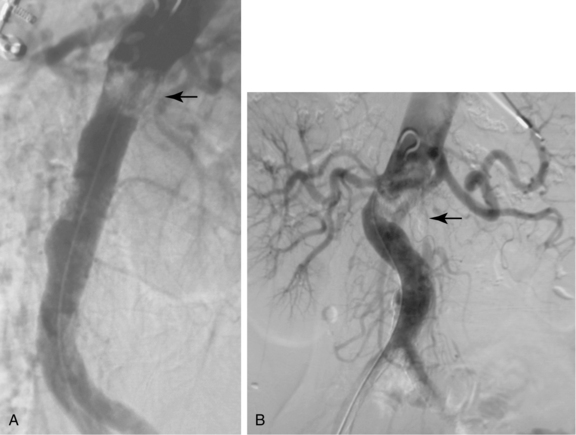
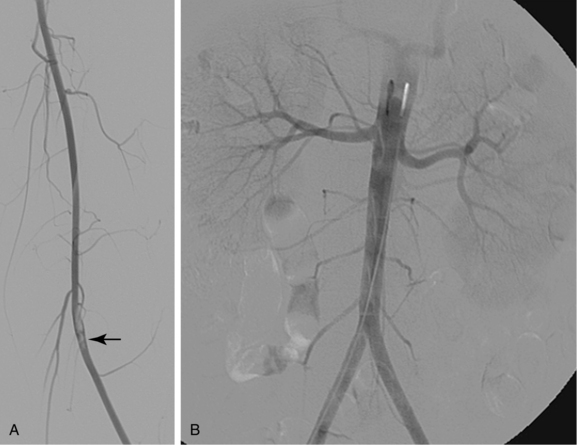
Atherosclerosis causes symptoms by blood flow reduction, thrombotic occlusion, plaque ulceration with distal embolization, and rarely by penetration into and through the media. Plaques can produce mild to severe irregularity of the wall or smooth, concentric narrowing (Fig. 1-7). A protruding plaque can mimic a luminal filling defect (Fig. 1-8). Significant atherosclerosis is most commonly seen at branch points and at certain anatomic sites, including the coronary arteries, carotid artery bifurcation, infrarenal abdominal aorta, and lower extremity arteries. Most affected patients have diffuse disease at many sites. Arterial luminal narrowing has several causes, although atherosclerosis is the most common (Box 1-2).
Neointimal hyperplasia and restenosis
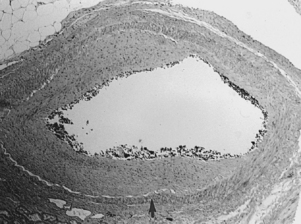
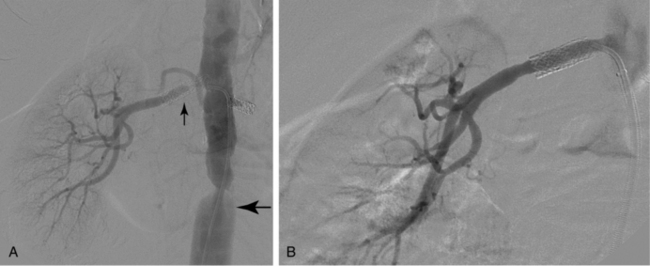
Neointimal hyperplasia is the “scar” produced by arteries (and veins) in response to significant injury or altered hemodynamics. Even though neointimal hyperplasia has features in common with atherosclerosis, it is a different pathophysiologic process. When caused by endovascular or surgical maneuvers (e.g., balloon angioplasty or stent placement), neointimal hyperplasia is triggered by clot formation and wall stretching at the site of injury.13 Over several days, monocytes and lymphocytes infiltrate the thrombus, which is itself partially resorbed. Growth factors released from smooth muscle cells, macrophages, and platelets cause smooth muscle cell proliferation and migration to form a thickened intima (Figs. 1-9 and 1-10). This evolution is complete within 3 to 6 months after injury.14 As with atherosclerosis, there is growing evidence that inflammation plays a central role in neointimal hyperplasia and restenosis.15,16
Thrombosis
The ingredients for thrombosis are platelets and other cellular blood elements, coagulation proteins, and often an abnormal endothelium. Clot formation begins with platelet adhesion and aggregation on the subendothelial vascular surface.17 Platelets release substances (e.g., adenosine diphosphate [ADP] and thromboxane A2) that further accelerate platelet activation. Aggregation of platelets occur by cross-linking of fibrinogen and von Willebrand factor through platelet surface αIIbβ3 integrin receptors (formerly designated as IIb/IIIa). Activation of the complex coagulation pathway (which begins with factor VII and tissue factor) leads to the formation of a “prothrombinase complex” (antiphospholipids bound to activated factors Va and Xa). Prothrombin is thus converted to thrombin, which is the critical enzyme responsible for transformation of fibrinogen to fibrin. A stable clot is formed from platelets, red blood cells, and white blood cells enmeshed within a fibrin matrix.
Classically, thrombosis occurs in the presence of vessel injury, slow flow, or a thrombophilic (hypercoagulable) state (i.e., Virchow triad). However, recent experimental work suggests that underlying inherited or secondary thrombophilia is the primary instigator of pathologic clot formation. In most cases, thrombi form at sites of preexisting disease (e.g., atherosclerosis, intimal hyperplasia) or acute trauma (see Figs. 1-2 and 1-7).
In addition to thrombosis, arterial occlusion has several other causes (Box 1-3).
Thrombophilia
A variety of hereditary and acquired disorders predispose to venous or arterial thrombosis17 (Boxes 1-4 and 1-5). Several inherited thrombophilias slightly to moderately increase the risk of pathologic thrombi. However, nontraumatic clot formation is much more likely in an individual when multiple congenital or secondary risk factors are present. Arterial thrombosis is particularly associated with antiphospholipid syndrome (APS), heparin-induced thrombocytopenia (HIT), protein C and S deficiencies, and myeloproliferative disorders (polycythemia vera and essential thrombocytosis.) Certain clinical situations should raise the suspicion for undiagnosed thrombophilia17 (Box 1-6).
Box 1-5 Secondary Thrombophilias
Box 1-6 Clinical and Laboratory Features of Primary Thrombophilias
Factor V Leiden refers to a mutation at the 1691 position of the gene coding for factor V. The activated cofactor is made resistant to activated protein C, thereby blunting its natural anticoagulant effect.18 Factor V Leiden is the most common inherited thrombophilia in Caucasian populations (about 5%). The lifetime risk for venous thromboembolic disease (VTE) for heterozygotes is increased 3- to 8-fold.19
Prothrombin gene mutation refers to a single point defect at the 20210 nucleotide position of the prothrombin gene that causes elevated levels of prothrombin in blood and renders the protein relatively resistant to APC. Like factor V Leiden, this inherited disorder only adds significantly to the risk of recurrent VTE when other hypercoagulable risk factors are also present.18–20
Antithrombin (AT) deficiency is a rare inherited autosomal dominant disorder expressed as lack of enzyme production (type I) or abnormal enzyme function (type II).21 AT is a key protein in regulation of coagulation by inactivation of thrombin and a variety of other coagulation factors. Although AT deficiency is a rare thrombophilia, it carries a 10- to 30-fold increased lifetime relative risk of venous (and less often arterial) thrombosis.18,19 An acquired form of AT deficiency is associated with liver disease and nephrotic syndrome.
Protein C and protein S deficiencies (as measured by activity or serum concentration) may be inherited or acquired. There is a 75% to 90% lifetime risk for VTE (and less often arterial thrombosis) in affected individuals with family members who have suffered a thrombotic event.19,22 Acquired protein C or S deficiency can be seen with septic shock, advanced liver disease, vitamin K antagonists (e.g., warfarin), HIV infection, nephrotic syndrome, acute inflammation, pregnancy, or oral contraceptive therapy.
Elevated coagulation factor levels are being recognized as a relatively frequent cause for apparently idiopathic thrombophilia. In particular, excessive levels of factors VIII, IX, and XI are associated with increased risk for VTE, although specific genetic abnormalities have not yet been isolated.23,24
Hyperhomocysteinemia refers to elevated blood levels of homocysteine, its oxidative metabolite homocystine, and other disulfides.17 A specific mutation in the gene for methylenetetrahydrofolate reductase (MTHFR) may cause mild hyperhomocysteinemia. When homocysteine levels are elevated, there is a significant added risk for venous or arterial thrombosis.25Homocystinuria is a rare autosomal recessive disorder caused by mutations of the cystathionine beta-synthase gene. The illness is marked by exceedingly high levels of homocysteine, arterial or venous thrombosis at an early age, premature atherosclerosis, Marfanoid features, and mental retardation.
Antiphospholipid syndrome (APS) is the most common acquired thrombophilia.17,26 The hallmarks of these conditions are apparently unprovoked vascular thrombosis and persistent elevation of antiphospholipid (aPL) antibodies of the lupus anticoagulant, anticardiolipin, or anti-beta-2-glycoprotein I type.27 The frequency of primary and secondary forms (the latter associated with connective tissue disorders such as systemic lupus erythematosus [SLE]) is about equal. The prototypical patient is a young to middle-aged woman with apparently idiopathic arterial or venous thrombosis. The risk for recurrent thrombotic events is significant. Because healthy people may transiently demonstrate aPL antibodies, positive clinical and laboratory criteria are needed for diagnosis.
Heparin-induced thrombocytopenia (HIT) is triggered by antibodies to heparin-platelet factor IV complexes in blood. These antibody complexes can precipitate platelet activation and degranulation, tissue factor and procoagulant release, and ultimately pathologic clot formation. About 3% of individuals who receive unfractionated heparin and 1.5% of those who receive low molecular weight heparin compounds will develop thrombocytopenia (<50% of baseline) within 4 to 10 days of the start of treatment in any form.28 Up to 50% of these patients will suffer HIT-related thrombosis (HITT), manifested by new or worsening VTE, arterial thrombosis, skin necrosis, or catheter-related deep vein thrombosis (DVT).29 When bleeding or thrombosis occurs, all heparin products must be stopped and replaced with a direct thrombin inhibitor.
Malignancy-associated hypercoagulability (once called Trousseau syndrome) is a multifactorial condition related to cellular release of procoagulants, tissue factors, cytokines, and platelet activators.30 Cancer is associated with impaired fibrinolysis, production of aPL antibodies, and acquired resistance to APC (Fig. 1-11). Thrombophilia is particularly common in several hematologic malignancies and in tumors of the pancreas, uterus, ovary, brain, stomach, lung, and prostate.17
Embolism
An embolus is any material that passes through the circulation and eventually lodges in a downstream vessel. Macroembolism and microembolism of the arterial circulation have numerous causes (Box 1-7).
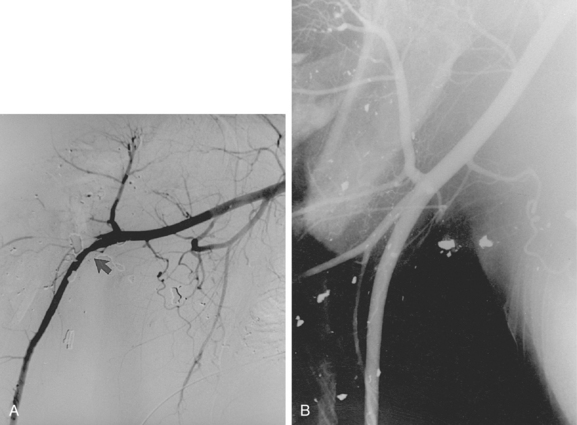
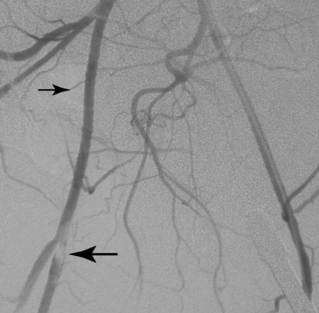
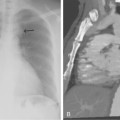
Macroemboli usually are clots that originate from the heart or a central artery. Atherosclerotic plaque also can fragment and obstruct peripheral arteries. Emboli tend to lodge at arterial bifurcations or at sites of preexisting disease. After the embolic event occurs, the distal arteries constrict, and new thrombus propagates proximally and distally to the level of the next large collateral branches. It may be impossible to differentiate thrombotic from embolic occlusions by imaging, although an acute embolus has several classic angiographic features (Box 1-8 and Fig. 1-12). Real or apparent luminal filling defects are also observed with intimal flaps, protruding atherosclerotic plaques, inflow defects, and rarely with intraluminal tumor (Fig. 1-13; see also Fig. 1-8). An arterial inflow defect is caused by unopacified blood entering an artery beyond an obstruction through a collateral vessel.
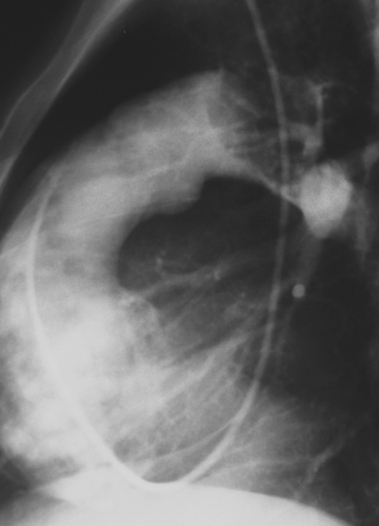
Microemboli are seen in patients with ulcerated, protruding atherosclerotic plaques. Platelet-fibrin deposits can be released spontaneously into the distal circulation from a site of underlying disease. Cholesterol crystals (100 to 200 μ) also may shower into the distal circulation from a plaque.31 Spontaneous release of small atheroemboli cause the blue toe (or blue finger) syndrome. However, the event may be associated with surgical manipulation, catheterization procedures, or treatment with anticoagulants or fibrinolytic agents.32 Widespread embolization into the legs, kidneys, head, or intestinal tract can result in acute renal failure, stroke, profound lower extremity or intestinal ischemia, and even death33 (see Chapter 2).
Aneurysms and arterial dilation
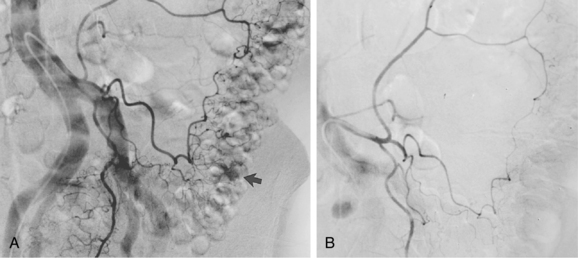
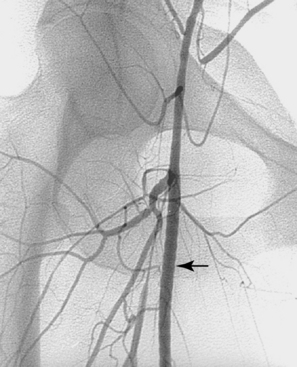
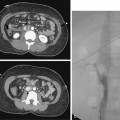
An aneurysm is defined as focal or diffuse dilation of an artery by more than 50% of its normal diameter.34 In a true aneurysm, all three layers of the arterial wall are dilated but remain intact (Fig. 1-14). Degenerative (atherosclerosis-associated) aneurysms fall into this category. In a false aneurysm (pseudoaneurysm), one or more layers of the arterial wall are disrupted (Fig. 1-15). Blood must be (temporarily) contained by the outer adventitia and surrounding supportive tissue. Trauma and infectious, neoplastic, or inflammatory masses typically produce pseudoaneurysms.35 True aneurysms usually are fusiform with diffuse dilation involving the entire circumference of an artery. False aneurysms often are saccular with focal, eccentric dilation involving part of the circumference of the vessel. However, these morphologic features are not always reliable for pathologic distinction.
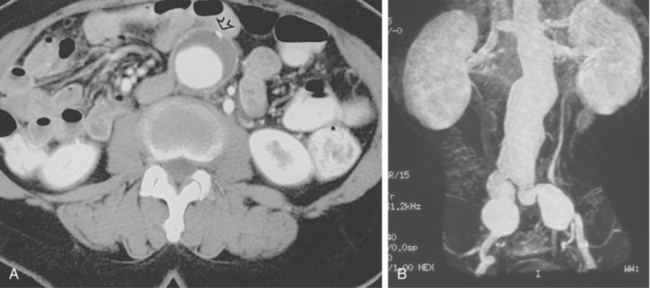
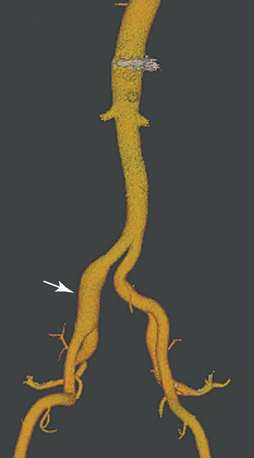
True and false aneurysms have a variety of causes (Box 1-9). Degenerative aneurysms are the most common. Although atherosclerosis and degenerative aneurysms are linked and often coexist in an individual, the two conditions are distinct disorders.36 Degenerative aneurysms form because of inflammatory damage to the vessel wall and hemodynamic forces that produce remodeling.37,38 The most common sites for degenerative aneurysms are the infrarenal abdominal aorta, descending thoracic aorta, and common iliac artery. Aneurysms of the popliteal, common femoral, internal iliac, brachiocephalic, and subclavian arteries are less common. Imaging features include diffuse arterial dilation, intimal calcification, and sometimes mural thrombus (see Fig. 1-14). The latter, which is common at most sites except the thoracic aorta, can obstruct branch vessels and give the lumen a smooth appearance.
Infectious (mycotic) aneurysms are caused by localized infection of the arterial wall.39,40 They occur after inoculation of a preexisting aneurysm or from infection and progressive dilation of a previously normal artery. The infection can arise through seeding of the artery from the lumen or vasa vasorum, invasion from a neighboring infection, or from penetrating trauma. Infectious aneurysms are typically saccular, occur at unusual sites, and can be multiple (see Figs. 6-28 and 7-25). They are most commonly seen in the aorta, viscera, and lower extremity arteries. In addition to bacterial infections, tuberculous arteritis may cause an aneurysm to form.41
Traumatic pseudoaneurysms follow blunt trauma (e.g., deceleration injury), criminal penetrating trauma, and medical procedures (e.g., catheterization, surgical repair) (see Fig. 6-21). Like infectious aneurysms, they usually are saccular and eccentric and often occur in the absence of other vascular disease. Other causes of aneurysms are considered in the following sections (Fig. 1-16).
Several forms of arterial dilation may be confused with an aneurysm:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


