Fig. 12.1
A T2w fast spin echo image of the liver demonstrating geographic variation of fibrosis. Clearly biopsies obtained at location 1 and 2 are likely to differ considerably in their grading undermining the concept of a whole liver fibrosis stage based on histopathology
12.4.2 CT Colonography and Optical Colonoscopy
Establishing the sensitivity of optical colonoscopy is an area that is difficult, largely because of the absence of an alternative, easily available reference standard. A per-polyp miss rate of 6 % for large adenomas (>10 mm) has been suggested by comparing back-to-back colonoscopies [56]. Given that this study used colonoscopy as its own reference standard, the miss rate of 6 % is likely to be an underestimate. For example, a polyp hidden behind a fold would be more likely to be missed by both colonoscopists (Fig. 12.2). This limitation may explain why when CT colonography is used as the reference standard, optical colonoscopy has a much higher miss rate of 12 % for large adenomas (10 mm or more in size) [51].

Fig. 12.2
A 1.5 cm polyp cancer at CT colonography on 2D (left) and 3D magnified (right). It is difficult to establish without repeat colonoscopy whether this polyp is actually a false negative at colonoscopy and true positive at CT colonography – or the reverse. It is typically very difficult to establish that a new test performs better than the reference test used and it has been argued that CT colonography should be the reference standard by which to validate optical colonoscopy
All colonoscopies are not equal however, for example, there is considerable variation in the polyp detection rate between colonoscopists [59] and even for the same colonoscopist at different times of day [66]. The upshot of this is that studies assessing the positive predictive value of CTC will be entirely dependent on the quality of local colonoscopy. Even in large academic centres, a significant proportion of so-called CTC false positives missed at initial colonoscopy will be true positive findings on repeat targeted colonoscopy [52].
12.4.3 CT Perfusion Metrics
As discussed earlier CT perfusion presents particular problems as none of the reference standards available in vivo can actually directly measure perfusion. Added to this the analysis models used for the biomarker itself vary, rely on different assumptions and algorithms (Fig. 12.3), may have different metrics and have been shown to generate differing results based on the same raw acquisition data [74]. In this situation it is clear that not only are the CT perfusion biomarkers generated by CT multiple but they have substantial intrinsic variability adding to the difficulty of validation.

Fig. 12.3
CT time-density curves (upper graph) from a dynamic liver study demonstrating the measured arterial signal (red) and the portal venous signal (blue). In the lower graph, a multi-parameter model has been used to fit a curve to the simultaneously acquired liver parenchyma signal measurements. Several different models have been used with varying background assumptions leading to variable results. In some applications image registration to counter physiological motion adds another variable factor into the analysis. The lack of a widely accepted robust reference standard for validating tissue perfusion biomarkers has probably contributed to the profusion of different types of model and analysis software, in turn limiting clinical application (Courtesy of Dr A. Gill)
PET-based radiotracer approaches offer possibly the best in vivo option for validating CT perfusion as a biomarker. However, these techniques also have several potential confounding factors, such as attenuation correction, low spatial resolution and correction for blood pool signal, and the results of the two techniques are difficult to directly compare. The modelling approaches taken for CT and PET perfusion have evolved separately and the metrics generated do not directly compare [1, 39]. In practice, the models developed for PET also differ between radiotracers. Although 15O-labelled water is an ideal tracer for flow quantification (as its uptake is linearly related to flow), a single-compartment model is used, whereas a 3-compartment model has been developed for blood flow quantification with 13N-ammonia and a two-compartment model is used for 82Rb [2].
12.5 Managing Imperfect Standards
Given that the majority of reference standards are imperfect, it is important that the limitations are recognised when validation is being considered. An important preliminary step is to ensure good reliability and repeatability of the biomarker itself. Then attempts should be made to improve the reliability and repeatability of the reference standard or develop a new more robust standard [20]. Typically, most reference standards are influenced by some factors which do not affect the biomarker so reliability and repeatability may be improved by identifying these factors and carefully controlling for them. This might be achieved by using carefully specified conditions for the validation process or by excluding those reference measurements where the additional factors clearly have an influence. If the standard remains “flawed” at this stage, it may still be possible to quantify any bias effect and account for this using a statistical model approach. Statistical strategies for combining information from two “imperfect” standards have been devised for the validation [42]. Alternatives might include the use of imaging “phantoms” where a carefully defined and known test object can be used as an ex vivo substitute for an in vivo measurement. This approach has been used, for example, to initially validate MR measurements of fat fraction, hepatic iron concentration and tissue stiffness; however ex vivo references usually introduce their own limitations, which may influence the results. Finally, where a suitable reference standard cannot be found to validate the biomarker, then it has been argued that it is still acceptable to provide specific qualification of a biomarker based on its performance against clinical endpoints.
12.5.1 Liver MRE and Histopathology
As discussed in the previous section, there are many reasons why histopathology is an imperfect measure of liver stiffness against which to validate MR elastography. The effects of some of these factors can be managed by controlling the relevant biomarker and reference standard conditions to make them both as likely as possible to reflect the desired underlying process. As an example for MRE, excluding livers from validation studies where there is known iron accumulation (Fig. 12.4) or regions of focal confluent fibrosis (on imaging) would reduce sampling variation. Avoiding cases with other obvious biomarker-confounding factors (Fig. 12.5) such as acute inflammation (e.g. acute hepatitis) or where elevated vascular or biliary tract pressure changes are likely (e.g. right heart failure, bile duct obstruction) could also improve the validation process. Using pathologist consensus and a single “fibrosis” grading scale may improve reliability and repeatability of the standard. Given the subjective and non-linear nature of the established grading scales for fibrosis, an alternative approach to improve the reference standard that has been tried is semiautomated quantitative collagen estimation [16, 46] within a selected microscope field of view. This is achieved using specific collagen stains such as sirius red and automated microscopy and image analysis methods to quantify the proportion of stain present as a percentage of the whole field of view [21]. Several authors have considered direct mechanical stiffness measurements in post-mortem livers, but clearly the lack of vascular perfusion and alteration of tissue mechanical properties with temperature make this approach unlikely to be comparable to in vivo measurements. A method for mechanically assessing the liver stiffness at open surgery has been developed [44], but as the authors discuss in some detail, their results do not correlate well with other approaches including MR methods. This is partly owing to their tube aspiration method being influenced by the constraining liver capsule. It is also not yet fully understood how liver stiffness is influenced by the “closed” abdomen and intra-abdominal pressure changes quite apart from possible influences of anaesthetic drugs on hepatic haemodynamics. Finally statistical approaches to utilising more than one imperfect reference standard have been applied to achieve liver fibrosis validation with mixed results [53, 54].


Fig. 12.4
An elastogram (left) giving heterogeneous values of 0–2kPa over the liver. This could be interpreted as normal, but in fact the MRE measurement has failed completely as liver signal is abnormally reduced on the T2w imaging (right) owing to accumulation of iron in the liver. This has reduced the signal from liver making shear wave detection impossible
Fig. 12.5
An abnormally stiff liver (>8kPa) at MRE indicating advanced fibrosis. However, it is important to recognise that increased liver stiffness may occur acutely owing to hepatitis, hepatic vein occlusion and biliary obstruction. Validation studies should recognise and exclude such confounding aetiologies for both the biomarker and the reference standard
12.5.2 CT Colonography and Optical Colonoscopy
Training is of crucial importance in maintaining a high adenoma detection rate, and improving training can have a positive effect [31]. In the UK, the Joint Advisory Group (JAG) on GI endoscopy sets standards for competence in GI endoscopy and quality assures endoscopy units, training and services. As an example of how JAG monitors standards, research using the JAG Endoscopy Training System (JETS) database has provided evidence that the current requirement for trainee colonoscopists to complete 200 procedures prior to assessment may be too few [70]. In the future, this research is likely to drive up the minimum requirements for independent practice.
Various technical improvements in colonoscopy have been used in attempts to drive up the adenoma detection rate. Chromoendoscopy describes the use of dye spraying during colonoscopy and is a way of improving sensitivity for diminutive polyps [33]. Technique changes, for example, changing the patient’s position during the examination [33], as well as the use of accessories such as the “Endocuff” [35], can also improve the adenoma detection rate.
In addition to improving the “gold standard”, the new biomarker can be validated against itself over time. For example, a polyp seen at CTC in the same place but larger over time is likely to be a genuine finding even if not seen at colonoscopy. This can be used as justification for repeating invasive tests such as colonoscopy [54].
12.5.3 CT Perfusion Metrics
CT perfusion biomarkers are largely based on pharmacokinetic models, and because there are several different models, different analysis software and potentially different methods of data acquisition, it is important to control for these. Most research groups have developed a specific technique often using a specific CT system to ensure acceptable repeatability and reliability of the biomarker metrics. It is of interest that the simplest least processed metric – the integrated area under the time-concentration curve (IAUCC) – is often the metric with highest correlation in many research studies rather than the model-based calculated parameters such as Ktrans, distribution volume, etc.
Having established a repeatable CT perfusion biomarker then, the problem of validation arises as the best techniques are not practical or ethical in humans in vivo. Animal-based validation has been performed successfully using microsphere techniques giving some confidence in the technique as discussed earlier, but in man, imperfect standards such as 15O or 82Rb tracer methods must be used. As these typically use different tracers (e.g. freely diffusible H2 15O vs extracellular iodine-based CT tracers), models and different metrics, direct comparison for validation is very difficult, and unsurprisingly results often vary substantially [22, 23]. In addition other factors may influence the reference standard results, for example, radioactive tracer dose variation, cardiac output, arterial blood sampling variation and filtering and post-processing algorithms. Instead of absolute metrics, those based on pre- and post-intervention ratio changes of the biomarker can be compared with the same changes in a reference standard, and this may prove to be the best in vivo validation that is available. As a result of these challenges and practical issues, the validation of CT perfusion in man has been severely limited. However, based on the evidence from animal work and clinical studies, and despite the relatively poor validation, there are many advocating [33] that the technique can still be successfully “qualified” for use as a tumour biomarker.
12.6 Biomarker Qualification
When a biomarker has been validated against a reference standard, it may still require qualification in order to confirm its relationship with all important clinical endpoints. This has become the accepted process for a biomarker to be approved by regulatory authorities such as the FDA or the EMA for use as a surrogate endpoint in clinical drug trials. Where validation of a biomarker proves impossible, for example, ethically, or is limited by the problems of the inadequate or impractical reference standard, then a case can still be made for biomarker qualification.
Qualification involves linking the biomarker directly to biological and clinical endpoints, and there is an inherent risk in this step as the plausible relationship between the biomarker and the underlying biology is extended towards disease outcomes and any assumed relationship may in fact be more associative than causal. The risk of confounding and bias increases and the biological linkage may no longer be valid – as in the CAST trial discussed initially. Qualification requires demonstrating that biomarker changes truly reflect clinical endpoints. This often requires carefully controlled conditions, for example, by limiting biomarker use to specific disease cohorts, specific clinical endpoints and under a specific range of conditions that reduces the risk of other factors influencing or confounding the results. This may involve constraining the biomarker acquisition to a specific imaging system manufacturer and analysis software. If prospective trials can demonstrate the value of the biomarker under these specific conditions, then it can be qualified for use in trials – but only under the same “qualifying” conditions.
12.6.1 Liver MRE
MRE has been extensively validated using histopathologic fibrosis stage as a gold standard, but awaits qualification for its use in predicting clinical endpoints such as development of HCC, liver decompensation or death. Given that the histopathologic fibrosis stage has only relatively recently been qualified against these clinical endpoints [14], it is perhaps not surprising that MRE awaits this step. There is some initial evidence that MRE-measured liver stiffness may be a risk factor for developing HCC in patients with chronic liver disease referred for MRI following an abnormal ultrasound [44]. Although this does not qualify the technique for the prediction of HCC development, it is at least encouraging. Further support can be drawn from qualification of similar biomarkers, in this case the demonstration that ultrasound elastography is predictive of clinical endpoints [50, 63], but this does not obviate the need for separate qualification of MRE. Several studies have also demonstrated that liver MRE could in future be qualified as a predictor of portal hypertension and variceal haemorrhage [59, 62, 67]. Given the limitations of histopathological staging, it is probably appropriate that qualification of hepatic MRE is explored until a better reference standard for validation is developed.
12.6.2 CT Colonography
CTC has been extensively validated in patients undergoing both CTC and optical colonoscopy (using optical colonoscopy as the gold standard) [3, 27, 30, 38]. Data to support qualification against hard clinical endpoints are emerging but as yet are incomplete. In patients undergoing CTC to clear the colon proximal to an obstructing tumour, there is evidence using the resection specimen as the gold standard that CTC is 100 % sensitive for malignancy but only 70 % sensitive for lesions ≥ 6 mm [51].
Qualification of CTC for its use more widely in symptomatic patients or as a screening tool is lacking. The endoscopic removal of colonic adenomas has been shown to reduce deaths from colorectal cancer [60]. The effect of carrying out CTC (and subsequent endoscopic polypectomy) on cancer-specific or all-cause mortality in symptomatic or asymptomatic patients has yet to be reported – this is likely a reflection of the relative novelty of the technique and the time it would take for meaningful results to emerge (i.e. at least 5 years). As there are ongoing trials evaluating CTC in screening, it may be that this is the next context for its qualification [11].
12.6.3 CT Perfusion Metrics
Owing to the difficulties of validating CT perfusion metrics in vivo in humans and the complexity of the various models and metrics employed, there has been a tendency to qualify CT perfusion metrics against clinical endpoints. Often these studies have involved relatively small cohorts and several authors [19] consider that further studies using much large cohorts are needed to properly qualify CT perfusion metrics for clinical use. In order to try and standardise as well as reduce the complexity and variability of CT perfusion metrics, several groups have also issued guidelines on the technique and analysis [41].
Many of these relatively small studies have demonstrated that CT perfusion metrics that indicate “high” levels of tumour perfusion at baseline correlate with a relatively good response to therapy and overall outcomes, for example, in breast cancer [37]. The survival and response of liver metastases to radioembolisation has been correlated with baseline CT perfusion metrics [43]. Perfusion metrics have also been used to predict survival in gliomas [73] and outcomes in head and neck tumours [5]. Other qualification studies include stroke outcomes which have been correlated with CT perfusion [52] and the risk of hyperperfusion syndrome after carotid stenting [75].
12.7 Pearls to Help Avoid Pitfalls
This chapter should make clear that full validation of imaging biomarkers against a robust reference standard is often extremely difficult to achieve. The list of key questions in Table 12.1 should be considered when validation of an imaging biomarker is being planned. It is important to understand that even following validation of a biomarker it is often impossible to demonstrate unequivocally that the underlying biological processes (and their reference standards) used for validation are causal in respect of the desired clinical endpoints when a biomarker is utilised in a specific disease setting. The inevitable desire to use convenient non-invasive imaging biomarkers as a surrogate endpoint in trials should not override the need for strong supporting evidence both for validation and qualification of the biomarker wherever possible. Many imaging biomarkers lack evidence in both respects but are being promoted as surrogates for use in clinical trials. The imaging community needs to be aware of the pitfalls not just inherent in the use of inadequate or misleading reference standards but perhaps more importantly of the need to focus carefully on undertaking carefully designed prospective qualification trials in ideally large populations. The paucity of these trials explains why, despite substantial interest in this area, only a small number of imaging biomarkers have survived the regulatory process of qualification as surrogate endpoints for drug trials or clinical use.
Table 12.1
 The Shift in Paradigm to Precision Medicine in Imaging: International Initiatives for the Promotion of Imaging Biomarkers
The Shift in Paradigm to Precision Medicine in Imaging: International Initiatives for the Promotion of Imaging Biomarkers
 Use Case II: Imaging Biomarkers and New Trends for Integrated Glioblastoma Management
Use Case II: Imaging Biomarkers and New Trends for Integrated Glioblastoma Management
 Introduction to the Stepwise Development of Imaging Biomarkers
Introduction to the Stepwise Development of Imaging Biomarkers
 A Proposed Imaging Biomarkers Analysis Platform Architecture for Integration in Clinics
A Proposed Imaging Biomarkers Analysis Platform Architecture for Integration in Clinics
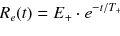 Imaging Biomarker Model-Based Analysis
Imaging Biomarker Model-Based Analysis
 Use Case III: Imaging Biomarkers in Breast Tumours. Development and Clinical Integration
Use Case III: Imaging Biomarkers in Breast Tumours. Development and Clinical Integration




Questions to ask before attempting imaging biomarker validation
Related posts:
The Shift in Paradigm to Precision Medicine in Imaging: International Initiatives for the Promotion of Imaging Biomarkers
Use Case II: Imaging Biomarkers and New Trends for Integrated Glioblastoma Management
Introduction to the Stepwise Development of Imaging Biomarkers
A Proposed Imaging Biomarkers Analysis Platform Architecture for Integration in Clinics
Use Case III: Imaging Biomarkers in Breast Tumours. Development and Clinical Integration
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
