Neurocutaneous syndromes encompasses a broad group of genetic disorders with different clinical, genetic, and pathologic features that share developmental lesions of the skin as well as central and peripheral nervous system. Cerebellar involvement has been shown in numerous types of neurocutaneous syndrome. It may help or be needed for the diagnosis and to explain the cognitive and behavioral phenotype of affected children. This article describes various types of neurocutaneous syndrome with cerebellar involvement. For each neurocutaneous disease or syndrome, clinical features, genetic, neuroimaging findings, and the potential role of the cerebellar involvement is discussed.
Key points
- •
Neurocutaneous syndromes are associated with widespread cerebellar involvement.
- •
Cerebellar involvement in certain types of neurocutaneous syndromes may cause neurocognitive deficits, in particular with regard to language and visuospatial abilities in children.
- •
Accurate characterization of cerebellar involvement may help in the diagnosis and influences long-term neurocognitive prognosis of children with neurocutaneous syndromes.
- •
In neurocutaneous disorder, cerebellar tumors such as medulloblastoma in basal cell nevus syndrome have a significantly different management regime compared with sporadic medulloblastoma.
Introduction
Neurocutaneous syndromes (NCS) are a group of congenital disorders of histogenesis in which the overall brain structure may be normal but anomalous cells persist and continue to differentiate. NCS primarily involves structures derived from the neuroectoderm and, consequently, typically affect the skin and central and/or peripheral nervous system. Most textbooks and reviews focus on the description of the typical supratentorial findings in NCS, with few focusing on the coexisting infratentorial lesions. Neuroimaging has proven to play a key role in the characterization, definition, and diagnosis of NCS. The cerebellum is, however, involved in various types of NCS and its careful evaluation should be part of every neuroimaging study in children with NCS. Cerebellar involvement may (1) be helpful or needed for the diagnosis of certain types of NCS and (2) explain the cognitive and behavioral phenotype (eg, impaired visuospatial ability, impaired language, or abnormal social behavior) of children with some NCS.
This article describes various types of NCS with cerebellar involvement. For each disease or syndrome, clinical features, genetic, neuroimaging findings, and the potential role of the cerebellar involvement is discussed.
Introduction
Neurocutaneous syndromes (NCS) are a group of congenital disorders of histogenesis in which the overall brain structure may be normal but anomalous cells persist and continue to differentiate. NCS primarily involves structures derived from the neuroectoderm and, consequently, typically affect the skin and central and/or peripheral nervous system. Most textbooks and reviews focus on the description of the typical supratentorial findings in NCS, with few focusing on the coexisting infratentorial lesions. Neuroimaging has proven to play a key role in the characterization, definition, and diagnosis of NCS. The cerebellum is, however, involved in various types of NCS and its careful evaluation should be part of every neuroimaging study in children with NCS. Cerebellar involvement may (1) be helpful or needed for the diagnosis of certain types of NCS and (2) explain the cognitive and behavioral phenotype (eg, impaired visuospatial ability, impaired language, or abnormal social behavior) of children with some NCS.
This article describes various types of NCS with cerebellar involvement. For each disease or syndrome, clinical features, genetic, neuroimaging findings, and the potential role of the cerebellar involvement is discussed.
Neurofibromatosis type 1
Neurofibromatosis type 1 (NF1; Online Mendelian Inheritance in Man [OMIM] entry 162200 ) is the most common NCS with a prevalence of 1 in 2500 to 3000 individuals. It is an autosomal dominant disorder caused by heterozygous mutation in the neurofibromin gene on chromosome 17q11.2. Neurofibromin is widely expressed with high levels in the nervous system and acts as a tumor suppressor. Neurofibromin reduces cell growth and proliferation by negative regulation of the cellular proto-oncogene p21RAS and by control of the serine threonine kinase mammalian target of rapamycin (mTOR). Impaired neurofibromin function predisposes to benign and malignant tumor formation.
The principal clinical manifestations of NF1 involve the skin and the nervous system, but the complications are variable and may involve most of the body systems.
Neuroimaging abnormalities in NF1 include intracranial neoplasms, parenchymal T2-hyperintense lesions, cerebral vasculopathy, and sphenoid wing dysplasia. Intracranial neoplasms include glioma, cranial nerve schwannoma, and plexiform neurofibroma. Gliomas generally develop in the optic pathways, brainstem, and, rarely, cerebellum. Optic pathway gliomas are the most frequent neoplasms seen in about 15% of children with NF1 and are typically low-grade pilocytic astrocytomas. Parenchymal T2-hyperintense lesions, also referred to as unidentified bright objects, can be seen in up to 75% of pediatric patients with NF1 and tend to decrease in prevalence with advancing age ( Fig. 1 A ). These lesions are not space occupying, do not or very rarely show contrast enhancement, and are typically located in the basal ganglia, internal capsule, brainstem, and cerebellum. Increased apparent diffusion coefficient (ADC) values in unidentified bright objects match the histopathological finding of myelin vacuolation and spongiotic changes attributed to increased water accumulation.
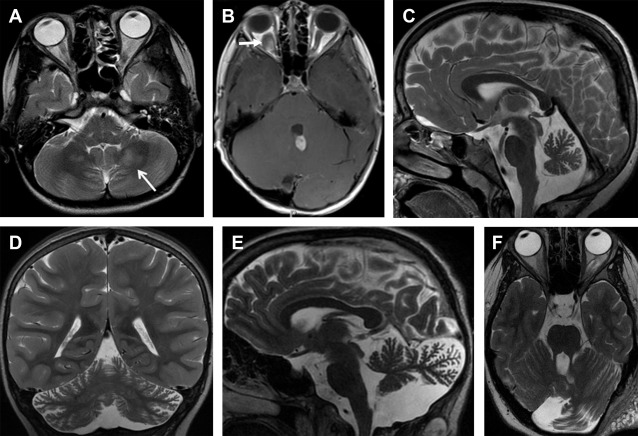
Primary cerebellar tumors are a rare presentation in NF1. In a series of 600 NF1 subjects, only 2 children had low-grade astrocytomas arising primarily from the cerebellar hemisphere ( Fig. 1 B). Vinchon and colleagues showed an overall better outcome for cerebellar gliomas associated with NF1 compared with sporadic cerebellar gliomas. Cerebellar parenchymal T2-hyperintense lesions can be distinguished from neoplasms based on absence of mass effect and lack of enhancement. Children with cerebellar parenchymal T2-hyperintense lesions showed a lower intelligence quotient and an altered cognitive profile compared with children without the lesions, particularly with regard to language and visuospatial abilities. Malformative cerebellar abnormalities, including cerebellar hypoplasia and dysmorphia (defined as enlargement of a cerebellar hemisphere with widening of the interfoliar spaces of its posterior part, which is bulky and crosses the midline), are a rare but most likely an underestimated cerebellar manifestation of NF1 ( Fig. 1 C–F).
Tuberous sclerosis complex
Tuberous sclerosis complex (TSC) is caused by either TSC1 (OMIM 191100 ) or TSC2 (OMIM 613254 ). It is an autosomal dominant NCS characterized by hamartomas in several organs, including the skin, brain, heart, eyes, kidney, lung, and liver. TSC is due to an inactivating mutation in 1 of the 2 genes, TSC1 (on chromosome 9q34) encoding hamartin or TSC2 (on chromosome 16p13.3) encoding tuberin. Hamartin and tuberin work together to inhibit the mTOR pathway that stimulates protein translation, cell growth, and proliferation. Mutations of hamartin or tuberin in TSC cause hyperactivation of the downstream mTOR pathway, which leads to disorganized cellular overgrowth, abnormal differentiation, increased protein translation, and the formation of tumors. Estimated prevalence of TSC is 1 in 6000 to 7000 newborns.
The identification of either a TSC1 or TSC2 pathogenic mutation in DNA from normal tissue is sufficient to make a definite diagnosis of TSC. Between 10% and 25% of TSC patients have no mutation identified by conventional genetic testing and a normal result does not exclude TSC or have any effect on the use of clinical diagnostic criteria to diagnose TSC.
Clinical features of TSC most commonly involve the brain (seizures, including infantile spasms, intellectual disability, autism, and self-injurious or aggressive behavior), skin (hypomelanotic macules, angiofibromas, ungula fibromas, shagreen patch, and confetti skin lesions), kidneys (angiomyolipomas and multiple renal cysts), heart (cardiac rhabdomyomas), eyes (multiple retinal hamartomas), and lungs (lymphangioleiomyomatosis in female patients). The 2012 International TSC Consensus Conference updated diagnostic clinical criteria now include 11 major features and 6 minor features. The 2012 consensus is based on definite (2 major features or 1 major feature with ≥2 minor features) or possible (either 1 major feature or ≥2 minor features) diagnosis. Interestingly, some neurologic symptoms such as seizures and intellectual disability are not part of the diagnostic criteria. Three major diagnostic criteria are based on neuroimaging studies (cortical dysplasias, including tubers and cerebral white matter radial migration lines [RMLs], subependymal nodules, and subependymal giant cell astrocytoma). This means that the diagnosis can be made by brain MR imaging alone.
The neuroimaging manifestations of TSC include cortical tubers, subependymal nodules, subependymal giant cell astrocytomas, and white matter RMLs. White matter RMLs are the most frequent neuroimaging finding and are strongly associated with age of seizure onset, intelligence outcomes, and level of autistic features. RMLs represent residual or altered heterotopic glial cells and neurons along the course of glial neuronal migration. Cortical tubers observed in approximately 90% of TSC patients are a type of focal cortical dysplasia and may occur in the cerebellum. The pathologic and clinical overlap between cortical tuber as a major feature and RML as a minor feature in the 1998 diagnostic criteria were replaced with a single major feature, cortical dysplasia, in the new 2012 classification.
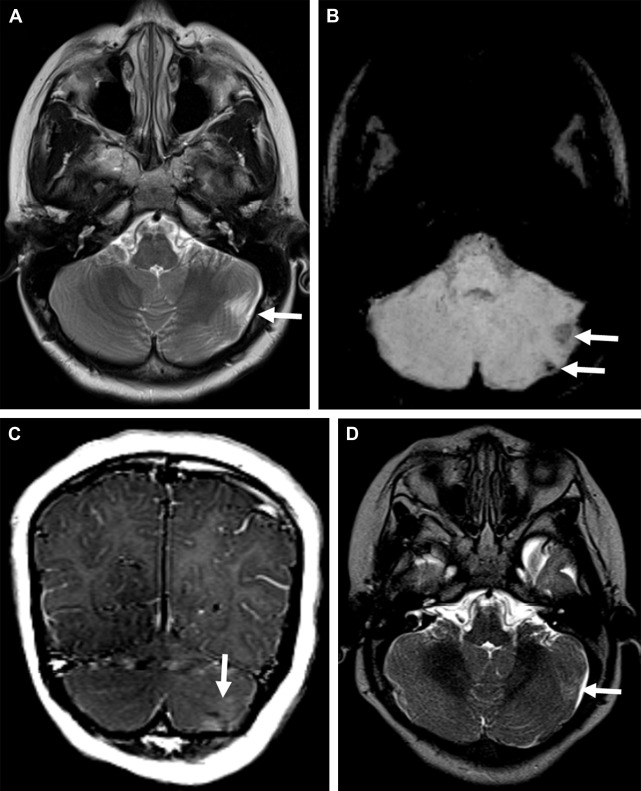
Cerebellar tubers are present in 24% to 36% of TSC patients and have a different imaging pattern compared with cerebral tubers. Cerebellar cortical tubers differ from the supratentorial tubers in their imaging features. They are triangular with a broad base towards the cerebellar cortex. The shape results from the differing neuronal migration within the cerebellum compared with the cerebrum. Cerebellar tubers are T1-hypointense, and T2-hyperintense ( Fig. 2 A ). Contrast enhancement with a striated or zebra-like pattern ( Fig. 2 B) and calcification ( Fig. 2 C) may be seen. Cerebellar tubers were demonstrated to increase in size, enhancement, or calcification, within the first 8 years of life ( Fig. 2 D). Elevated ADC values in cerebellar tubers have been attributed to gliosis and hypomyelination. An increasing number of cerebellar tubers on MR imaging has been correlated with the severity of autistic spectrum disorder. In addition, reduced cerebellar volume has been observed in TSC patients, particularly patients with TSC2 mutations and a more severe phenotype. Evidence of the role of cerebellar involvement in cognition and behavior in TSC was provided by animal studies. In a Tsc1 mouse model, Tsai and colleagues showed a decrease in the number of Purkinje cells compared with control. This is the most likely explanation for smaller cerebellar volume in TSC patients. In addition, mutated mice showed an autistic-like behavior as seen in TSC patients.
Sturge-Weber syndrome
Sturge-Weber syndrome (SWS; OMIM 185300 ) is a sporadic NCS characterized by a facial cutaneous capillary malformation (port-wine stain) in the ophthalmic distribution of the trigeminal nerve, ipsilateral vascular glaucoma, vascular malformation of the choroid, and a concomitant vascular malformation of the brain and meninges (leptomeningeal angioma). The estimated prevalence of SWS ranges between 1 in 20,000 to 50,000 live births. Specific somatic mosaic activating mutation in GNAQ is associated with both SWS and nonsyndromic port-wine stains. It is hypothesized that GNAQ mutation causes dysregulation of vascular endothelin that may result in malformed, progressively dilated, and abnormally innervated blood vessels. Pathophysiological mechanism in SWS can be explained by venous dysplasia producing focal venous hypertension and resultant tissue hypertrophy. The absence of a mature venous system is initially compensated by the persistence and enlargement of the primitive primordial cerebral venous system. This primitive venous system becomes progressively insufficient with the rapid brain development resulting in intraparenchymal venous hypertension, venous ischemia, tissue injury, and atrophy. Compensatory collateral venous circulatory pathways (eg, dilated intramedullary veins) draining into the deep venous system partially compensate for the venous insufficiency. The clinical course of SWS is variable but typically includes seizures, hemiparesis, headache, stroke-like episodes, developmental delay, and visual field defects.
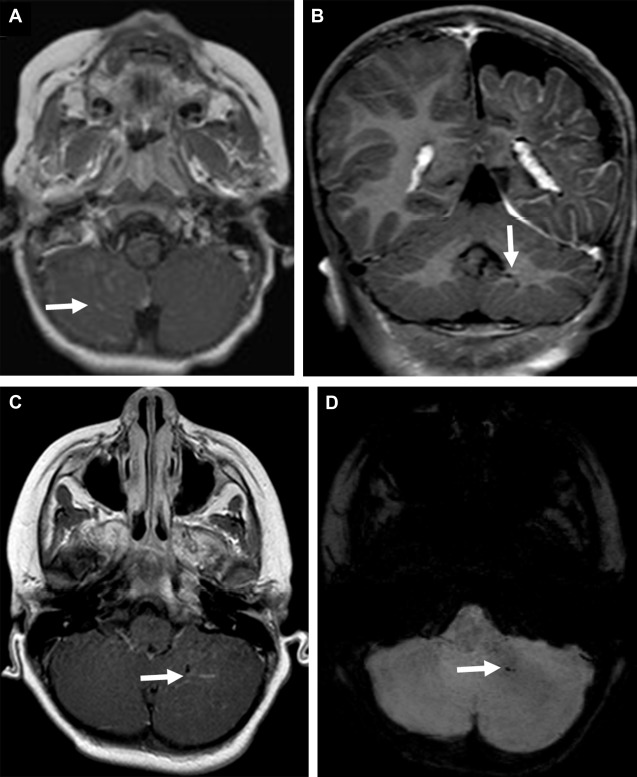
Typical neuroimaging findings in SWS include a prominent leptomeningeal angiomatosis (especially well seen on contrast-enhanced T1-weighted and FLAIR images), dilated medullary veins, enlargement of the ipsilateral choroid plexus, and underlying slowly progressive cortical or subcortical atrophy. The occipital lobes are primarily affected; in severe cases the central zone, as well as frontal lobes, may also be involved. Rare cases of bilateral involvement have been recorded. Susceptibility-weighted imaging (SWI) is an advanced MR imaging technique that can detect deoxygenated blood in small veins without contrast administration. SWI can detect dilation of transmedullary veins secondary to venous hypertension. In addition, progressive cortical calcifications secondary to long-standing venous hypertension or ischemia is easily identified by the T2* blooming artifacts on SWI.
Cerebellar involvement in SWS is rare and includes leptomeningeal enhancement ( Fig. 3 A ), atrophy, and developmental venous anomaly ( Fig. 3 B–D). ADC-values of the cerebellar white matter remote from the location of the leptomeningeal angioma may be increased.
PHACE syndrome
PHACE syndrome (OMIM 606519 ) is an NCS characterized by posterior fossa malformation, infantile hemangioma (IH), arterial anomalies, coarctation of the aorta, eye abnormalities, and ventral developmental defects, specifically sternal defects, and/or supraumbilical raphe. The cause and pathogenesis of PHACE is unknown and its diagnosis is based on the presence of a characteristic hemangioma and other major or minor clinical or imaging criteria.
Isolated IH are subtle or absent at birth, usually becoming more evident within the first days to weeks of life. IH associated with PHACE tend to be large (>5 cm in diameter), telangiectatic in appearance, and typically segmental in distribution in the face. Most patients have a normal neurologic examination in infancy but may develop focal seizures, developmental delay, and recurrent headaches. Early neurologic findings are typically related to structural brain anomalies.
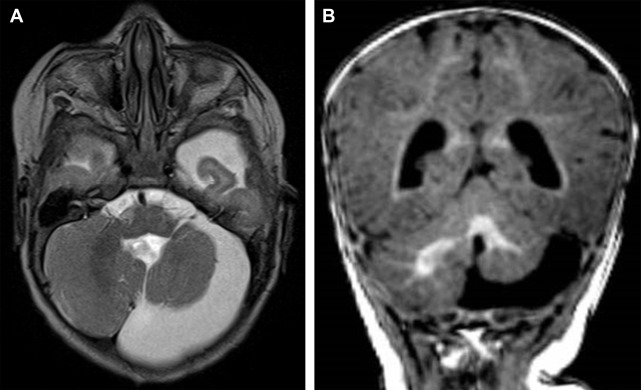
The presence of a segmental cervicofacial IH should prompt neuroimaging to evaluate for the presence of intracranial abnormalities that may be related to PHACE. Hence, neuroimaging plays a key role in establishing the diagnosis. Intracranial anomalies (cerebrovascular and posterior fossa) are the most common extracutaneous feature of PHACE and are typically ipsilateral to the IH. Intracranial and cervical arteriopathy diagnosed by MR angiography can be categorized as dysgenesis, narrowing, nonvisualization, persistent embryonic carotid-vertebrobasilar arterial connections, and abnormalities in arterial course and/or origin. Arterial dysgenesis is the most common vascular manifestation and is characterized by eccentric outpouching, fusiform enlargement, or aneurysm formation. Arteriopathy is more common in the anterior circulation (internal carotid artery) than in the posterior circulation. Perinatal arterial ischemic stroke may be seen. Supratentorial brain abnormalities associated with PHACE include callosal dysgenesis, hemispheric hypoplasia, subependymal heterotopia, polymicrogyria, and extra-axial hemangiomas. Posterior fossa anomalies are the most commonly reported brain anomalies in PHACE. The typical posterior fossa anomaly is unilateral cerebellar hypoplasia ( Fig. 4 ) with or without involvement of the vermis; whereas a Dandy-Walker malformation (DWM) is less common.
von Hippel-Lindau disease
von Hippel-Lindau (VHL) disease (OMIM 193300 ) is an autosomal dominant NCS characterized by various benign and malignant tumors of the central nervous system (CNS), kidneys, adrenal glands, inner ear, and reproductive adnexal organs such as retinal, cerebellar, and spinal hemangioblastoma and endolymphatic sac tumor. VHL results from a germline mutation in the VHL tumor suppressor gene on chromosome 3p25.3, which inactivates a VHL allele, with the carriers of this mutation being subject to a second inactivating event (2-hit hypothesis). This mutation results in complete functional loss of the tumor suppressor and, therefore, overexpression of proteins that mediate angiogenesis. The estimated incidence of VHL is about 1 in 36,000 live births. Outside the CNS, multisystem visceral involvement includes renal cysts, renal cell carcinomas, pancreatic cysts, and pheochromocytomas. CNS hemangioblastoma is observed in 65% of patients with VHL and is a defining feature in VHL.
The cerebellum is the most common location (about 65%) for CNS hemangioblastomas. They are highly vascular benign World Health Organization (WHO) grade 1 tumors that show solid enhancement, appear pial-based, and are, therefore, peripherally located in the cerebellum ( Fig. 5 ). When the tumors enlarge, they appear cystic with an enhancing mural nodule and the natural history of progression shows a faster rate of cystic expansion than growth of the causative solid tumor, eventually resulting in pressure effect and clinical symptoms. Hemangioblastomas are T1-isointense and T2-hyperintense. Heterogeneous T2 signal may be seen in the presence of intralesional hemorrhage. Mural nodules are well demarcated and show homogenous enhancement. If present, flow voids appear T2 hypointense with superficial heterogeneous enhancement. Peritumoral vasogenic edema is typically mild. In VHL, multiple lesions may occur simultaneously. Additional lesions may be present along the spinal cord. Imaging of the entire neural axis is consequently advised. The lesion may mimic a pilocytic astrocytoma. The clinical findings usually allow differentiation. In addition, hemangioblastomas are typically characterized by the prominent vascular signal voids. Finally, hemangioblastomas may hemorrhage into the subarachnoid space with resultant hemosiderosis due to their high vascular nature and close proximity to the subarachnoid space. Endolymphatic sac tumors are, with a prevalence of up to 16%, a component of VHL disease. Contrast-enhanced MR imaging of the petrous bone must be part of annual surveillance.
Related posts:
 The Role of the Pediatric Cerebellum in Motor Functions, Cognition, and Behavior
Cerebellar and Brainstem Malformations
The Pediatric Cerebellum in Inherited Neurodegenerative Disorders
The Role of the Pediatric Cerebellum in Motor Functions, Cognition, and Behavior
Cerebellar and Brainstem Malformations
The Pediatric Cerebellum in Inherited Neurodegenerative Disorders
 Normal Cerebellar Development by Qualitative and Quantitative MR Imaging
Normal Cerebellar Development by Qualitative and Quantitative MR Imaging
 Prenatal Cerebellar Disruptions
Prenatal Cerebellar Disruptions
 Vascular Disorders of the Cerebellum in Children
Vascular Disorders of the Cerebellum in Children
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


