Grade
Mitotic per 10 high-power microscopic fields (HPF)
Proliferative index/Ki-67 index (%)
Low grade (G1)
<2
≤2
Intermediate grade (G2)
2–20
3–20
High grade (G3)
>20
>20
Aim of Surgery
Surgery aims either to provide/improve local control of disease burden, or to stop the natural course of the disease and, above all, definitively cure the patient. Treatment strategy and surgical indications are highly variable according to tumor site, genetic origin, local and regional extension, and biological behavior of the tumor.
Despite lack of high-level evidence such as randomized controlled trials, surgical strategies for NET are now better defined. Most of these recommendations are detailed in the European Neuroendocrine Tumor Society (ENETS) guidelines recently published and available online (www.enets.org). If some surgical indications are consensual, surgery should also sometimes be avoided.
Overall, surgical resection is rarely an “oncological emergency,” and a temporary “wait and see” policy is often acceptable to better assess the tumor natural history. If some surgical indications are consensual, surgery should also sometimes be avoided. Considering the rarity of such disease, surgery needs to be discussed on a case-by-case basis in a multidisciplinary neuroendocrine tumor board.
When Surgery is Required
First, it is noteworthy that surgery is the single most effective therapy for NET. Whatever abdominal procedure is planned, cholecystectomy should always be performed during primary resection of the tumor [32]. This is justified by the risk of gallstone-related complications or acute cholecystitis if patients are later treated with somatostatin analogs or liver arterial embolization [33, 34].
Some surgical indications are consensual, because of the clear benefit on long-term outcome.
Gastric Neuroendocrine Tumor
Gastric NETs represent less than 10 % of digestive NET. They can be due to chronically elevated gastrin (ECLomas) because of achlorydia from atrophic fundic gastritis (type 1, the most frequent) or to gastrin tumoral secretion (type 2, associated with Zollinger–Ellison Syndrome—ZES) (Table 6.2). Patients with type 1 or type 2 gastric NET above 1 cm with deep gastric parietal wall invasion and/or positive margins after endoscopic resection should undergo surgical resection [35–37]. It is important to recognize the low malignancy risk of type 1 lesions. These lesions can be treated either by local resection or antrectomy (by open or laparoscopic approach) [38], and this latter anatomical resection can, in theory, suppress the source of gastrin and decrease recurrence rate [35]. Overall, total gastrectomy should be avoided when possible, and its indications are limited to widely diffuse and large or malignant lesions. Whatever procedure performed, endoscopic surveillance is recommended thereafter [39].
Table 6.2
Gastric neuroendocrine tumor: risk of malignancy, surgical indication, and disease-specific mortality
Tumor type | Risk of malignancy | Surgical indication | Disease-specific mortality (%) |
|---|---|---|---|
Type 1 | Very low (2–5 %) | Surgery if >1 cm, not endoscopically resectable, deep parietal invasion (beyond submucosa), nodal or distant spread | ±0 |
Type 2 | Low (10 %) | Local excision surgery if large or positive margins | <10 |
Type 3 | High (>50 %) | Formal gastrectomy in all cases | 25–30 |
Regarding rare sporadic primary gastric NET (type 3), they carry an overall dismal prognosis and should undergo curative-intent (R0) resection, by formal gastrectomy according to tumor location with regional lymphadenectomy, similar to gastric adenocarcinoma.
Duodenopancreatic Neuroendocrine Tumor
Insulinomas are the most common functioning endocrine neoplasms of the pancreas, frequently presenting with nonspecific symptoms due to hypoglycemia, such as weakness, confusion, headaches, sweating, tremors, palpitation, and visual disturbances. They are most of the time sporadic and benign, with less than 5 % of them being associated with MEN 1 and less than 10 % being malignant (i.e., with distant or nodal metastasis) [40, 41]. Accurate preoperative localization and characterization (including endoscopic ultrasonography (EUS) [42, 43], triphasic CT scan, or MRI [44]) are mandatory in order to tailor surgical treatment and to avoid blind distal pancreatectomy as it used to be recommended. Usually, allow adequate tumor localization and can avoid invasive exams such as intra-arterial calcium stimulation with hepatic venous sampling [44, 45]. Additionally, EUS can accurately assess relationship between the tumor and the main pancreatic duct. Surgery is the treatment of choice with an overall cure rate close to 100 % for benign lesions, if complete resection is achieved [46]. Procedures should be performed by an experienced team in pancreatic surgery and can be either performed laparoscopically or through open approach [41, 47]. After additional intraoperative localization of the tumor by palpation and ultrasonography, the lesion can be either enucleated or resected by standard pancreatectomy such as pancreaticoduodenectomy or distal pancreatectomy. Whenever possible, parenchyma-sparing resection [48] including central pancreatectomy or enucleation should be preferred because of a better long-term exocrine and endocrine function. Enucleation is best indicated for small benign lesions located in the head of the pancreas and far enough, i.e., 2–3 mm, from the main pancreatic duct [49]. Interestingly, preoperative EUS can accurately help the surgeon to choose the adequate surgical procedure assessing relationship between the tumor and the main pancreatic duct.
In patients with MEN 1, insulinoma can be multiple in about 10 % of patients and one should keep in mind of it in order to locate all lesions, pre and/or intraoperatively, thus avoiding blind pancreatectomy.
Treatment for other rare duodenopancreatic secreting lesions is in first line surgical resection [50], as for sporadic gastrinoma, glucagonoma, or vipoma. Most of these lesions are malignant, and standard pancreatectomy with formal lymphadenectomy is required. Regarding duodenal gastrinoma that even when malignant, often grows slowly, routine duodenotomy during surgical exploration usually allows accurate identification, and consequently, local resection can be performed. It is important to note that local lymphadenectomy should be systematically performed in order to decrease recurrence rate. Nodal extension of disease can occur in about 45 % of both duodenal and pancreatic gastrinomas [51].
Nonsecreting tumors began to be more incidentally diagnosed because of the widespread use of cross-sectional imaging, representing up to 75 % in recent surgical series [52, 53]. Their natural history is heterogeneous and difficult to assess during preoperative workup, and whether an incidental finding is associated with improved prognosis is still a matter of debate [52, 53]. Nevertheless, size being an important prognostic factor, surgical resection with regional lymphadenectomy is required for lesions above 2 cm [52]. Indeed, in this setting, the risk to develop metastatic disease during the follow-up is above 10 % [54].
Small Bowel Neuroendocrine Tumor
They represent from 25–40 % [55–57] of gastrointestinal NET. They mostly occur after 60 years of age, can be multiple in up to 40 % of cases, and they present with carcinoid syndrome in about a quarter of patients [55, 57]. Their prognosis is not as good as for other gastrointestinal tumors justifying an aggressive surgical management. However, the overall survival is around 60 % and can be up to 85 % in cases with curative resection [17, 58]. If possible, primary should be localized before surgery, using double balloon enteroscopy, capsule endoscopy, CT/MRI enterography as well as cross-sectional, and nuclear imaging such as 18F-DOPA-PET.
Even small and asymptomatic lesions need to undergo surgery because lesion size does not correlate with biological behavior, and they can be associated with node or liver metastases [15]. Small bowel lesions need to go through segmental resection with a formal wide lymphadenectomy including all gross metastatic nodes even when they are located around the superior mesenteric artery origin. Nevertheless, a special attention must be paid to avoid large resections leading to small bowel syndrome. Surgery for the primary tumor should always be considered even in the presence of metastatic disease, since the primary lesion can be responsible for local complications such as intussusception, small bowel obstruction, or ischemia. Additionally, also some lesions can present with important peritumoral fibrosis involving the mesentery root and the retroperitoneum, leading to occlusion, hydronephrosis, and chronic pain. Studies showed better results among patients who had at least their primary tumor resected along with nodal resection, with a better disease-free and overall survival [15, 17]. Since abdominal complications remain one of the major causes of death, we believe that in the setting of unresectable lesion, 90 % cytoreductive surgery can be considered, if a long enough small bowel can be conserved [56]. Surgery is at best performed after medical control of carcinoid syndrome, if present, by somatostatin analogs. Preoperatively, a special attention must be paid to carcinoid heart disease assessment in case of patient with carcinoid syndrome. Up to now, laparoscopic approach in this setting has been poorly studied, but we believe that open approach should be preferred in order to explore the full length of small bowel and to achieve a large lymphadenectomy.
Despite complete surgical resection of small bowel carcinoids, recurrence can occur in about 30–40 % of cases [16, 59]. Liver recurrence is best treated with resection in the case it is possible, which occurs usually in less than 20 %. Otherwise, other modalities of local treatment such as chemoembolization and pure embolization can be used as well as systemic therapy, from somatostatin analogs to chemotherapy.
Appendiceal Neuroendocrine Tumor
Appendiceal NET is currently diagnosed incidentally after appendectomy. They represent about a third of all gastrointestinal endocrine tumors and are the most benign of carcinoids. Although the overall 5-year survival is around 85 %, size is the most important prognostic factor. Tumors below 1 cm are cured by appendectomy alone, without need for any additional treatment. For tumors above 2 cm, a right colectomy is needed [60], due to the risk of lymph node extension. Between 1 and 2 cm, the risk-to-benefit ratio needs to be determined on a case-by-case basis.
Colonic and Rectal Neuroendocrine Tumor
These rare tumors are often incidentally diagnosed during colonoscopy and are nonfunctioning in most of the cases.
For well-differentiated colonic tumors, colonic resection with standard oncologic criteria should be performed in a similar way to adenocarcinoma, because of the poor 5-year prognosis of these tumors, between 40 and 70 %.
For well-differentiated rectal tumors below 1 cm, developed within the submucosal layer (T1) and without nodal involvement on preoperative EUS and MRI, local resection, either endoscopical or surgical is appropriate. For tumors above 2 cm, standard oncologic anterior resection is required. Between 1 and 2 cm, the risk-to-benefit ratio needs to be determined on a case-by-case basis.
Concerning endoscopic resection, endoscopic submucosal resection has replaced standard polypectomy as the preferred technique for tumors below 1 cm, and newer techniques such as submucosal resection with band ligation or endoscopic submucosal dissection are likely to be associated with less residual disease [61]. Submucosal resection with band ligation has the advantages of being easier to perform, demanding a shorter procedure time and having better negative margin rates; thus, it may be considered the treatment of choice for small rectal carcinoid tumors [62, 63].
Neuroendocrine Tumor Within a Genetic Background
NET can be associated with multiple endocrine neoplasia type 1, or less frequently with von Hippel–Lindau’s disease, von Recklinghausen’s disease, or tuberous sclerosis (Table 6.3) [64].
Table 6.3
Frequency and type of pancreatic neuroendocrine tumors in patients with genetic syndrome
Genetic syndrome | Frequency of pancreatic neuroendocrine tumors (%) | Types of pancreatic neuroendocrine tumor |
|---|---|---|
MEN 1 | 80–100 | Nonfunctioning tumors, gastrinomas, insulinomas |
VHL | 12–20 | Usually nonfunctioning tumors |
NF-1 (von Recklinghausen) | <10 | Mainly somatostatinoma; gastrinomas, insulinomas, and nonfunctioning tumors |
Tuberous sclerosis | <5 | Nonfunctioning tumors, gastrinoma, insulinoma |
Regarding MEN 1, tumors such as nonfunctioning tumors above 2 cm [65], glucagonoma, vipoma, somatostatinoma, or GRFoma should undergo standard resection, since malignant NET is one of the main determinants of long-term survival in this disease. Insulinoma [66] represents a formal indication of resection because of its potentially harmful secretion (Table 6.4) [67].
Table 6.4
Types of tumor and prevalence in MEN type 1 patients
Type of tumor | Prevalence (%) |
|---|---|
Nonfunctioning | 80–100 % (microadenomas); 55–80 % (>1 cm) |
Gastrinomas | 25–50 |
Insulinomas | 20 |
Glucagonomas | 3 |
Vipomas | 1 |
Somatostatinomas | 1 |
GRHomas | 1 |
In VHL disease, pancreatic NETs are usually nonfunctioning and have a better prognosis than sporadic nonfunctioning pancreatic NETS (metastatic disease in 10–20 vs. 60–90 %), and patients are usually younger about 30 years [68–70]. Tumors can also be multiple in about 20 % and locate throughout the pancreas. Usually, nonsurgical management is recommended for those patients, but criteria predicting poor prognosis have been described, being: tumor size ≥3 cm, tumor doubling time ≤500 days, and mutation in exon 3. In the case that more than two of these criteria are present, surgery is to be considered. Otherwise, surveillance with CT/MRI can be advocated [68, 69]. Patients with VHL should be screened for pancreatic lesions starting at 12 years of age and resecting PNETs should be considered if the patient is having an exploratory laparotomy for another manifestation of VHL [70].
NETs related to NF-1 are usually somatostatinomas frequently located around the papilla. These tumors metastasize in one-third to half of cases irrespective of primary tumor size. Although some experts advocate local excision for tumors smaller than 2 cm and surgical excision for tumors larger than 2 cm, as many as 70 % of surgical cases are associated with regional or liver metastasis, an aggressive surgical approach is best indicated in the form of pancreaticoduodenectomy [70–73].
When Surgery Should be Avoided
Surgery should only be avoided after a complete clinical, biological, and imaging workup and discussion in a multidisciplinary neuroendocrine tumor board. Workup should include complete clinical examination, static and dynamic biological tests adapted to tumor origin and secretion. Imaging workup should at least include thoraco-abdominal CT scan, to assess tumor burden. When liver is involved, because of the frequent numerous small metastases [75], a liver MRI should be added routinely. If in doubt, nuclear imaging should be performed to better assess tumor biology (18F-FDG-PET CT) or tumor burden (111In-somatostatin receptor scintigraphy, 68GA-DOTATOC PET, or 18F-DOPA-PET).
Gastric Neuroendocrine Tumor
In patients with gastric NET associated with chronic atrophic gastritis (type 1) or with Zollinger–Ellison syndrome with multiple endocrine neoplasia type 1 (type 2), tumors under 1 cm can undergo endoscopic resection and follow-up with yearly endoscopic surveillance (every 6–12 months) with gastric biopsies is necessary [35–37], because of their very low risk of lymph node extent.
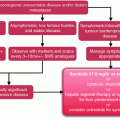
High-grade and Poorly Differentiated Tumors
These tumors are characterized by a mitotic count over 20 per high-power field and/or a Ki-67 index over 20 % and a poorly differentiated pathologic pattern. These tumors have a specific and very aggressive biological behavior totally different from G1 and G2 tumors. Platin-based chemotherapy is the first line treatment for these tumors. Surgical indications are very limited and can be considered for very localized tumors and/or tumors well controlled under chemotherapy.
When Surgery Should be Discussed
Metastastic Tumor to the Liver
The presence of metastases, most commonly located in the liver, is a major adverse prognostic factor [76–78] and affects up to 75 % of patients. Metastases are often bilobar, numerous, and synchronous. Despite a lack of high-level evidence, complete (R0) resection is the only hope for cure in these patients, and an aggressive surgical approach—including resection of metastatic liver disease—is widely accepted [2–18]. Nevertheless, intra- and extrahepatic recurrences are frequent, and a careful patient selection is needed. Patient selection is based on operative risk and general status, but mainly on tumor biological behavior. Surgery should be only discussed for patients with well-differentiated tumor (G1 and G2), at low risk of postoperative mortality and with no extrahepatic disease, when R0 resection is technically doable. On a technical point of view, two-step hepatectomy or intraoperative ablation enables complete resection with low mortality in patients with bilobar liver metastases, with a 5-year overall survival above 90 % and a 5-year disease-free survival around 50 % [79]. It is important to note that metastases are sometimes far more numerous than what is previously suggested by imaging [75].
Unresectability is defined as the technical impossibility to achieve R0 resection: metastases involving the right or left hepatic pedicle and abutting the contralateral pedicle, or involving or abutting the vena cava, or involving two hepatic veins and abutting the third one, or lesions that would leave <25 % of functional liver after resection. It is important to note that unresectability should be only defined par an experienced team including a hepatobiliary surgeon. Whether, in this setting, debulking surgery is justified remains highly controversial and should be limited to highly selected cases if at least 90 % of the tumor could be resected.
A recent meta-analysis demonstrated that surgery was superior to chemoembolization or other treatment modalities (non surgical) in the treatment for liver metastases of NET, with a significant longer survival [80].
Orthotopic liver transplantation has been proposed as an alternative option. Indeed, liver metastases are one of the main causes of dead, are usually confined to the liver for a long time, and can be responsible of debilitating symptoms.
Main criteria for transplantation are the absence of extrahepatic disease, a low Ki-67, and symptomatic disease refractory to previous therapies. In a French multicentric study, overall 5-year survival was 50 % and poor prognostic factors included upper-abdominal exenteration, primary tumor in duodenum or pancreas and hepatomegaly [81]. A review including 150 transplanted patients extracted from the UNOS database [82] demonstrated a long-term survival similar to that of patients with hepatocellular carcinoma, especially in patients with stabilized disease. Additionally, a recent European multicentric study of 213 patients showed an overall survival of up to 60 % after liver transplantation and suggested hepatomegaly, concurrent resections and age over 45 years as poor outcome predictors in the more recent cases [83]. Nguyen et al. also showed 5-year survival rate of near 60 % after 2002 and introduction of the Model for End-Stage Liver Disease (MELD) criteria [84]. Máthé et al. [85] found that age over 55 years and simultaneous pancreatic resection were poor prognostic factors in a study with 89 patients (Table 6.5).
Table 6.5
Survival after liver transplantation for neuroendocrine tumors