CHAPTER 35 Posterior Fossa Intra-Axial Tumors
Brain parenchyma of the posterior fossa includes the cerebellar hemispheres, vermis, pons, midbrain, and medulla oblongata. Neoplasms arising from these structures are a significant source of morbidity and mortality, especially in the pediatric patient. A clear understanding of the tumors that present in the posterior fossa is essential for the radiologist performing brain imaging in these patients.
MEDULLOBLASTOMA
Epidemiology
Medulloblastoma is primarily a tumor of young children, but it has been reported in all age ranges. It is the most common malignant brain tumor diagnosed in children and is the most commonly encountered of the pediatric posterior fossa tumors. It accounts for up to 25% of all pediatric brain tumors and close to 40% of posterior fossa pediatric brain tumors. Medulloblastoma is slightly more common in boys and is infrequently diagnosed in adults. When considering only pediatric patients, the mean age at diagnosis is just over 7 years. However, the distribution can be parsed further, with a typical presentation occurring in children 1 to 4 years of age and a variant group presenting at 6 to 9 years of age. Recent population studies have identified increased rates of diagnosis associated with birth in the autumn and winter and increased number of siblings.1
Clinical Presentation
The characteristic presentation of medulloblastoma is that of an infant or preschool-aged child with signs and symptoms of increased intracranial pressure caused by obstruction of the ventricular system at the level of the fourth ventricle. Such signs and symptoms include headache, irritability, nausea/vomiting, papilledema, and macrocephaly.1 Symptoms caused by local effects of the tumor in the posterior fossa include cranial nerve palsies and ataxia. Vomiting may be the result of increased intracranial pressure or direct impingement of the tumor on the area postrema on the floor of the fourth ventricle. Less common presenting symptoms include neurologic deficits associated with spinal cord compression from metastatic disease, seizure, or precipitous neurologic decline due to intratumoral hemorrhage. Symptoms are generally short in duration before diagnosis, reflecting the rapid growth of the tumor.
Pathophysiology
Over a third of medulloblastoma patients will have loss of genetic material from chromosome 17. Genetic defects have also been identified in chromosomes 1, 8, 9, 10, 11, and 16.2 Nevoid basal cell carcinoma syndrome (NBCCS, Gorlin’s syndrome), an autosomal dominant–inherited cancer syndrome caused by genetic mutations in the PTCH gene on chromosome 9, is associated with an increased risk for the development of medulloblastoma. Other clinical features of the syndrome that can be identified on imaging studies include odontogenic keratocysts of the jaw and calcifications of the falx cerebri. The latter may be present at a much earlier age than typical senescent meningeal calcifications. Medulloblastoma has also been reported in Li-Fraumeni syndrome, Rubenstein-Taybi syndrome, and Turcot’s syndrome.3
Pathology
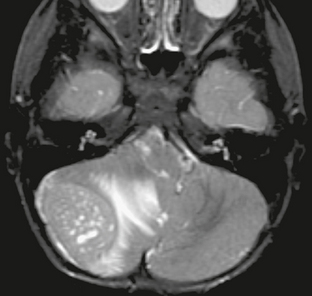
Medulloblastoma is characterized as a grade IV tumor by the World Health Organization (WHO), with four major subtypes. The most commonly encountered subtype is classic medulloblastoma, characterized by monotonous sheets of round cells with abundant nuclei and scant cytoplasm (Fig. 35-1). The cells may form Homer-Wright rosettes, with tumor cells radially arranged around fibrillary processes. The desmoplastic medulloblastoma contains “pale islands” of cells with abundant neoplasm, surrounded by more primitive-appearing tumor cells. Medulloblastoma with extensive nodularity and advanced neuronal differentiation (nodular medulloblastoma) is almost exclusively diagnosed in children younger than 3 years of age, with tumor cells clustered in nodules surrounded by a fibrillary matrix. This histologic subtype is frequently identifiable by its clinical imaging appearance (Fig. 35-2). The least common of the four major subtypes is the large cell medulloblastoma, which contains large cells with abundant neoplasm and prominent nucleoli.4,5
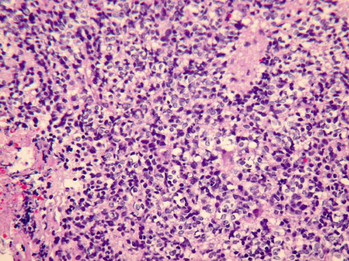
FIGURE 35-1 Medium-power photomicrograph of medulloblastoma demonstrates monotonous sheets of small cells with scant cytoplasm and multiple mitotic figures (H&E stain).
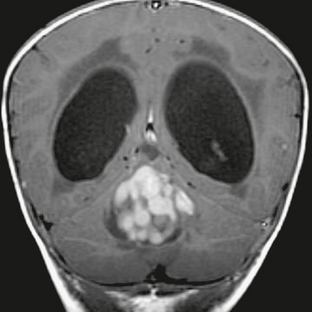
FIGURE 35-2 Coronal T1W MR image after contrast agent administration shows the grape-like clusters of solid enhancing tumor characteristic of nodular medulloblastoma.
Imaging
CT
CT is often the first diagnostic imaging study performed on children subsequently diagnosed with medulloblastoma, and specific features on CT can increase confidence in the preoperative diagnosis. Unusual among posterior fossa tumors in children, solid portions of medulloblastoma are characteristically hyperattenuating at CT before contrast agent administration (Fig. 35-3); most other posterior fossa tumors of childhood are slightly hypoattenuating. This CT imaging feature is thought to reflect the high nuclear-to-cytoplasmic ratio and dense cellularity of the tumor. Up to 20% of medulloblastomas will demonstrate some calcification at CT, but this is not a helpful differentiating finding. However, if calcifications are identified along the falx cerebri, a diagnosis of Gorlin’s syndrome should be strongly considered. Although cyst formation is more typically associated with cerebellar astrocytoma, it is common to identify small cysts or regions of necrosis within a medulloblastoma; these may be more apparent on MRI. Also more apparent on MRI is the presence of vasogenic edema surrounding the tumor. Tumors that are primarily within the fourth ventricle will often elicit little or no surrounding vasogenic edema, but this is not a reliable distinguishing feature. Advanced CT techniques, including CT angiography and CT perfusion, have not been extensively applied in the evaluation of medulloblastoma.
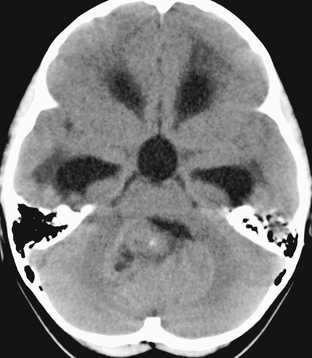
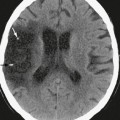
FIGURE 35-3 Axial noncontrast CT of a 2-year-old with medulloblastoma. The tumor is isodense to gray matter in the cerebellar hemispheres, with a small focus of calcification anteriorly. It causes obstructive hydrocephalus, with periventricular interstitial edema evident at the margins of the frontal and temporal horns.
MRI
MRI is the diagnostic study of choice in evaluation of medulloblastoma, allowing for multiplanar analysis and sensitive detection of characteristic features of the tumor itself and of metastatic lesions in the brain and spinal canal. The same features that cause high attenuation on CT can result in solid portions of the tumor appearing isointense relative to gray matter on T2-weighted (T2W) and fluid-attenuated inversion recovery (FLAIR) images. However, these sequences also emphasize small cystic foci within the tumor that are hyperintense relative to cerebrospinal fluid (CSF) within the ventricles. In addition, these tumors will appear relatively hyperintense juxtaposed to the middle cerebellar peduncles bordering the fourth ventricle (Fig. 35-4). Thus, the concept of the “dark on T2” medulloblastoma is misleading; it is more accurate to say that these tumors tend to be hypointense on T2W images relative to other pediatric posterior fossa tumors. Gradient-echo imaging will increase sensitivity for the detection of hemorrhage or calcifications, but hemorrhagic foci within these tumors are rare, and the detection of calcification is of little help in narrowing the differential diagnosis. On T1-weighted (T1W) images, medulloblastomas are typically isointense to hypointense, exhibiting variable degrees of enhancement after contrast agent administration.
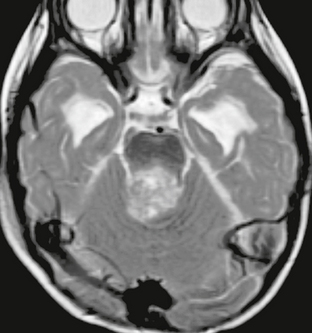
FIGURE 35-4 Axial T2W MR image of medulloblastoma filling the superior aspect of the fourth ventricle. Although solid portions of the tumor are isointense relative to gray matter, the overall appearance is that of a hyperintense mass.
Upward of a third of children with medulloblastoma will have imaging evidence of subarachnoid spread of tumor at the time of diagnosis.1,6 Although contrast enhancement is the primary means of identifying subarachnoid spread of tumor both in the cranial vault and in the spine, it should be recognized that some metastatic lesions will exhibit little or no enhancement. Contrast-enhanced sagittal images can often clearly demonstrate the base of the tumor arising from the “roof” of the fourth ventricle. This can be a key diagnostic clue to the correct diagnosis (Fig. 35-5); the less common ependymoma is much more likely to arise from the “floor” of the fourth ventricle, growing dorsally. Dynamic MR perfusion imaging can demonstrate increases in blood flow associated with angiogenesis within high-grade tumors. Application of this imaging technique in the evaluation of medulloblastoma has been sporadic, and its impact on clinical decision-making in this diagnosis remains uncertain.
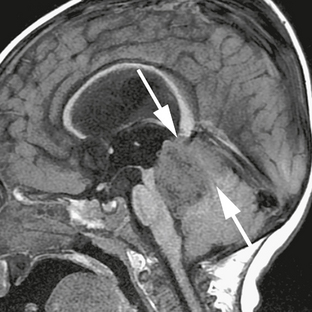
FIGURE 35-5 Sagittal noncontrast T1W MR image showing the poor definition of the junction of tumor with the roof of the fourth ventricle (arrows), indicating the superior medullary velum as the site of origin of this medulloblastoma. The margin between the tumor and the floor of the fourth ventricle is much more clearly defined.
Diffusion-weighted imaging (DWI) characteristically demonstrates restricted diffusion within medulloblastomas (Fig. 35-6); and when compared with other intra-axial posterior fossa tumors in children, medulloblastoma shows a consistently greater degree of diffusion restriction.7 In one study,8 an apparent diffusion coefficient (ADC) value of less than 0.9 × 103 mm2/s was specific for medulloblastoma, relative to cerebellar astrocytoma and ependymoma.
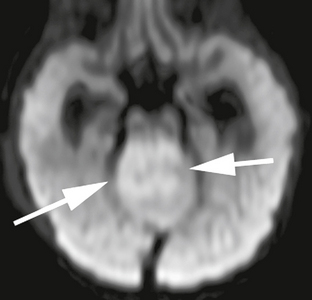
FIGURE 35-6 DWI demonstrates restricted diffusion in the solid portions of this medulloblastoma (arrows).
MR spectroscopy is frequently employed in the preoperative evaluation of medulloblastoma. These tumors show a characteristically aggressive metabolite spectrum, with marked elevation of choline and suppression/absence of N-acetyl-aspartate (NAA) (Fig. 35-7). Lipid and lactate elevation can be seen as a manifestation of anaerobic metabolism in the tumor. Several authors have reported frequent elevation of taurine, at 3.3 ppm.9 The anatomy of the posterior fossa may lead to some degradation of spectroscopic quality, but multi-voxel spectroscopic imaging can help distinguish laterally based tumors (Fig. 35-8) from regions of adjacent edematous brain.
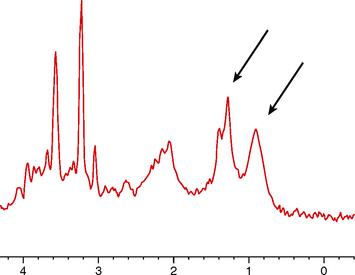
FIGURE 35-7 MR spectroscopy of medulloblastoma. Arrows point to the resonance doublet of lipid and lactate, reflecting the anaerobic metabolism characteristic of this aggressive tumor.
EPENDYMOMA
Pathophysiology
In general, ependymomas are relatively well-differentiated tumors with multiple ultrastructural features. This accounts for their heterogeneous appearance on imaging and at gross inspection, as well as their relatively slow growth. The majority of posterior fossa ependymomas are classified as WHO grade II lesions. A small proportion of ependymomas will exhibit more aggressive features at histologic analysis, with dense cellularity and prominent mitotic activity; these are termed anaplastic ependymomas.
Multiple chromosomal aberrations have been identified in association with ependymoma, including defects on chromosomes 1, 6, 9, 13, 16, 17, 19, 20, and 22.10 There is a high incidence of spinal ependymomas in neurofibromatosis 2.
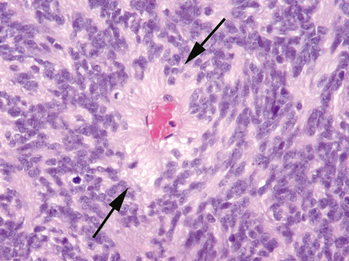
Pathology
Four pathologic subtypes of ependymoma have been described. The classic ependymoma is characterized by a moderately cellular and lobular pattern of growth interrupted by perivascular pseudorosettes (Fig. 35-9). These structures demonstrate strong immunoreactivity for glial fibrillary acidic protein (GFAP). True ependymal rosettes can also be identified, characterized by columnar epithelium surrounding a distinct lumen. The rare tumors with a growth pattern resulting in extensive and complex epithelial surfaces have been termed papillary ependymomas. Another uncommon subtype is characterized by cells that have a prominent perinuclear halo and nuclear uniformity, called clear cell ependymoma. The fourth variant of ependymoma encountered in the posterior fossa in children is the tanycytic ependymoma, in which individual cells are markedly elongated with fibular processes resembling pilocytic astrocytoma. The familiar myxopapillary ependymoma is essentially restricted to an intraspinal location at the filum terminale.4
Imaging
CT
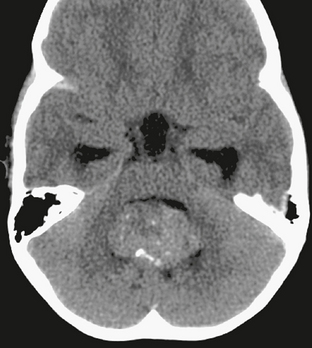
CT of posterior fossa ependymoma typically demonstrates a heterogeneous tumor filling and expanding the fourth ventricle, with extension laterally through the foramen of Luschka. Close to 50% of posterior fossa ependymomas will demonstrate calcification on CT (Fig. 35-10), and many will show regions of hemorrhage or necrosis. Enhancement is variable and irregular.
MRI
The MRI appearance of posterior fossa ependymoma reflects the heterogeneity of the tumor. T2W images demonstrate a tumor with solid components that are isointense or hyperintense to brain parenchyma, markedly hypointense regions reflecting hemorrhage and/or calcification, and cystic foci. A similar pattern is seen with FLAIR sequences (Fig. 35-11), with pronounced hyperintense signal in cystic or necrotic regions. Hemorrhagic and calcified areas may demonstrate hyperintense signal on T1W images, and the heterogeneous and variable pattern of contrast enhancement exhibited on CT is duplicated with MRI. As with medulloblastoma, sagittal postcontrast imaging can be very helpful in demonstrating point of attachment of an intraventricular tumor (Fig. 35-12). When this point of attachment is from the dorsal brain stem with tumor growth into the ventricle itself, a diagnosis of ependymoma or exophytic brain stem glioma is likely.
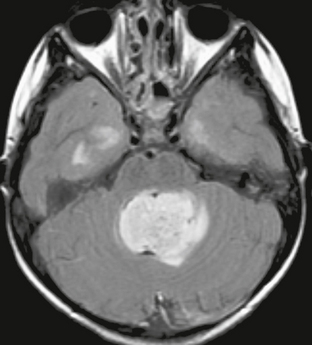
FIGURE 35-11 Axial FLAIR image of a fourth ventricular ependymoma in a 7-year-old. The tumor elicits little edema in the adjacent cerebellum.
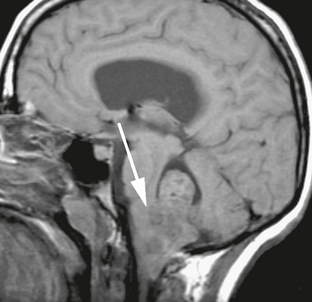
FIGURE 35-12 Sagittal noncontrast T1W MR image of the fourth ventricular ependymoma with inferior extension through foramen magnum. Arrow points to site of attachment of tumor to the dorsal inferior aspect of the brain stem.
DWI demonstrates restricted diffusion in infratentorial ependymoma but not as pronounced as in the more common medulloblastoma. The degree of restriction is typically greater than in pilocytic astrocytoma,8 and DWI will also show restricted diffusion in areas of necrosis.