Chapter 7 Primary Mediastinal Neoplasms
Introduction
Neoplasms of the mediastinum are a diverse group of benign and malignant pathologic entities that include both primary and secondary malignancies. Most neoplasms of the mediastinum are metastases, typically from lung cancer, although extrathoracic neoplasms such as breast cancer and melanoma have a predilection for spread to the mediastinum. Primary neoplasms of the mediastinum are uncommon, and whereas the majority in adults are benign, those in children are malignant.1 In terms of primary neoplasms, the most common anterior mediastinal neoplasms include thymomas, teratomas, and lymphomas. Neoplasms of the middle mediastinum are typically congenital cysts, including foregut and pericardial cysts, whereas those that arise in the posterior mediastinum are often neurogenic neoplasms.
Epidemiology and Risk Factors
Thymic, germ cell, and neurogenic neoplasms are a heterogeneous group of primary mediastinal neoplasms. Thymic neoplasms account for 17%, lymphomas 16%, and neurogenic neoplasms 14% of all cases of primary mediastinal neoplasms.2 Germ cell tumors (teratomas, seminomas, embryonal carcinomas, endodermal sinus tumors, and choriocarcinomas) account for approximately 15% of all mediastinal tumors in adults and 24% in children.3 The remainder of the neoplasms in the mediastinum represents a large group of miscellaneous entities that includes mediastinal cysts (bronchogenic, esophageal, pericardial, thymic, and neurenteric) that account for 15% to 20% of all mediastinal masses.
Thymomas are the most common of the thymic neoplasms and characteristically are located in the anterior mediastinum. Thymomas typically occur in patients older than 40 years, are rare in children, and affect men and women equally.4–6 Thymic neuroendocrine tumors (carcinoid, small cell carcinoma, and large cell carcinoma) are uncommon. Thymic carcinoid tumor is the most common of this group of tumors. Affected patients are typically in the fourth and fifth decades of life and there is a male predominance.
Germ cell tumors usually occur in young adults (mean age 27 yr).7 Most malignant germ cell neoplasms (>90%) occur in men, whereas benign neoplasms (mature teratomas) occur with equal incidence in men and women.7 Teratomas are the most common germ cell neoplasm, representing 70% of all germ cell neoplasms in childhood and 60% in adults.7 Men and woman are equally affected. Malignant germ cell neoplasms are divided into seminomas and nonseminomatous neoplasms. Seminomas are the most common pure histologic type, accounting for 40% of such neoplasms, and usually occur in men between the third and fourth decades of life.7 The nonseminomatous germ cell tumors of the mediastinum include embryonal cell carcinoma, endodermal sinus tumor, choriocarcinoma, and mixed germ cell tumors. Teratoma with embryonal cell carcinoma (teratocarcinoma) is the most common subtype, whereas pure endodermal sinus tumors, choriocarcinomas, and embryonal carcinomas are much less common.
Neurogenic neoplasms represent 75% of all primary posterior mediastinal masses.8 These neoplasms are classified as tumors of peripheral nerves (neurofibromas, schwannomas, and malignant tumors of nerve sheath origin), sympathetic ganglia (ganglioneuromas, ganglioneuroblastomas, and neuroblastomas), or parasympathetic ganglia (paraganglioma and pheochromocytoma). Peripheral nerves are more commonly involved in adults and schwannomas constitute 75% of this group, whereas sympathetic ganglia neoplasms are more common in children. Schwannomas and neurofibromas typically occur with equal frequency in men and women, most commonly in the third and fourth decades of life. Thirty percent to 45% of neurofibromas9 occur in patients with neurofibromatosis (NF), and multiple neurogenic tumors or a single plexiform neurofibroma is considered pathognomonic of the disease.10 Malignant tumors of nerve sheath origin (also termed malignant neurofibromas, malignant schwannomas, or neurofibrosarcomas) are rare and typically develop from solitary or plexiform neurofibromas in the third to fifth decades of life. Up to 50% occur in patients with NF-1, and in these patients, tumors occur at an earlier age (typically adolescents) and with a higher incidence than in the general population.4,11 Neuroblastomas are the most common extracranial solid neoplasms in children, accounting for 10% of all childhood neoplasms. Neuroblastomas are typically diagnosed at a median age of younger than 2 years, ganglioneuroblastomas at 5.5 years of age, and ganglioneuromas at 10 years.12
Anatomy and Pathology
The anterior mediastinum is bounded anteriorly by the sternum; posteriorly by the pericardium, aorta, and brachiocephalic vessels; superiorly by the thoracic inlet; and inferiorly by the diaphragm. Its contents include the thymus, lymph nodes, and fat. The middle mediastinum is bounded by the pericardium, and its contents include the heart and pericardium, aorta, vena cava, brachiocephalic vessels, pulmonary vessels, trachea and main bronchi, lymph nodes, and phrenic, vagus, and left recurrent laryngeal nerves. The posterior mediastinum is bounded anteriorly by the trachea and pericardium, anteroinferiorly by the diaphragm, posteriorly by the vertebral column, and superiorly by the thoracic inlet. Although the true anatomic posterior boundary is the vertebral column, with respect to mediastinal disease, masses in the paravertebral regions are usually included in the posterior mediastinum. The contents of the posterior mediastinum include the esophagus, descending aorta, azygos and hemiazygos veins, thoracic duct, vagus and splanchnic nerves, lymph nodes, and fat.13
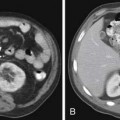
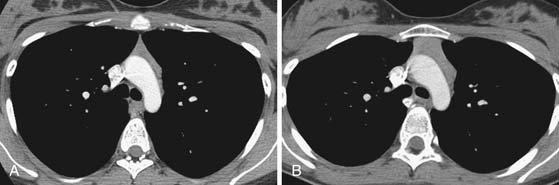
In the anterior mediastinum, significant proportions of the neoplasms arise from the thymus. Accordingly, an understanding of the normal appearance and location of the thymus is important in the detection and diagnosis of these neoplasms. The thymus consists of two lobes placed in close contact along the midline extending from the fourth costal cartilage upward, as high as the lower border of the thyroid gland. However, ectopic thymic tissue can occur at any level in the pathway of normal thymic descent, from the angle of the mandible to the upper anterior mediastinum. The two lobes generally differ in size; the left lobe is larger than the right and extends more inferiorly. Thymic hyperplasia, an increase in weight and size, has two distinct histologic forms: true and lymphoid hyperplasia. True hyperplasia occurs in children and young adults recovering from severe illness or trauma or after chemotherapy (Figure 7-1). Lymphoid hyperplasia occurs most commonly in patients with myasthenia gravis but also in association with other diseases such as hyperthyroidism, Graves’ disease, rheumatoid arthritis, and scleroderma.14
Thymic neoplasms are usually epithelial in origin and include thymomas and carcinomas. Most thymomas are solid neoplasms that are encapsulated and localized to the thymus. However, one third have necrosis, hemorrhage, or cystic components; invasion of the capsule and involvement of the surrounding structures occurs in approximately one third of cases.15 Thymomas have a wide variety of histologic features, and there is a strong association between the histologic findings and invasiveness as well as prognosis.16 Thymic carcinomas usually manifest histologically as large, solid, and infiltrating masses with cystic and necrotic areas. They are histologically classified as low or high grade, with squamous cell–like or lymphoepithelioma-like variants being the most common cell types.4 Thymic neuroendocrine neoplasms are uncommon. These tumors typically manifest as a large, lobulated, and usually invasive anterior mediastinal mass that can exhibit areas of hemorrhage and necrosis.4,17 Malignant potential ranges from relatively benign (thymic carcinoid) to highly malignant (small cell/large cell carcinoma of the thymus). Typical carcinoid has low mitotic activity (<2 mitoses/2 mm2) without necrosis, whereas atypical carcinoid has a higher degree of mitotic rate (2-10 mitoses/2 mm2) and/or necrosis. Small cell and large cell neuroendocrine carcinomas have a higher rate of mitotic activity (>10 mitosis/2 mm2) and associated necrosis.
In the posterior mediastinum, most of the neoplasms are neurogenic in origin. Nerve sheath tumors are either schwannomas, neurofibromas, or malignant tumors of nerve sheath origin (malignant neurofibroma, malignant schwannoma, and neurogenic fibrosarcoma). Histologically, schwannomas are encapsulated tumors which arise from Schwann cells located in the nerve sheath and grow along the nerve, causing extrinsic compression.11 They are heterogeneous in composition and can have low cellularity, areas of cystic degeneration, hemorrhage, and small calcifications. Neurofibromas differ from schwannomas in that they are unencapsulated and result from proliferation of all nerve elements, including Schwann cells, nerve fibers, and fibroblasts. They grow by diffusely expanding the nerve. Plexiform neurofibromas are variants of neurofibromas that infiltrate along nerve trunks or plexuses.18 Ganglion cell tumors arise from the autonomic nervous system rather than nerve sheaths and range from benign encapsulated neoplasms (ganglioneuromas) to moderately aggressive neoplasms (ganglioneuroblastomas) to malignant unencapsulated masses (neuroblastomas). They are derived from cells of embryologic origin or sympathetic ganglia. After the abdomen, the thorax is the second most common location of neuroblastomas,19 whereas ganglioneuromas and ganglioneuroblastomas are more common in the sympathetic chain of the posterior mediastinum. Ganglioneuromas are benign tumors composed of one or more mature ganglionic cells. Ganglioneuroblastomas have histologic features of both ganglioneuromas and neuroblastomas. They are the least common type of neurogenic tumors. Neuroblastomas are the most aggressive type and are composed of small round cells arranged in sheets or pseudorosettes.20,21
Key Points Anatomy: Anterior mediastinal masses
• Thymic origin: Thymic hyperplasia, thymic epithelial tumor (thymoma, thymic carcinoma, thymic carcinoid, thymolipoma), and cyst.
• Germ cell tumors: Teratoma (mature, immature, malignant), seminoma, nonseminomatous.
• Lymphoma: Hodgkin’s and non-Hodgkin’s.
• Thyroid mass: Goiter, thyroid cancer.
• Miscellaneous: Adenopathy, parathyroid adenoma, mesenchymal tumors (lymphangioma, hemangioma).
Key Points Pathology
• Thymic neoplasms: Epithelial (thymomas and carcinoma); neuroendocrine tumors (carcinoid-typical and atypical, small and large cell neuroendocrine carcinoma).
• Germ cell tumors: Teratoma (mature, immature, and malignant); nonteratomatous tumors (seminomas and nonseminomas). Seminomas (germinomas); nonseminomatous germ cell tumors (embryonal carcinoma, endodermal sinus tumor, choriocarcinoma, and mixed type).
• Neurogenic sheath tumors: Schwannomas or neurofibromas.
• Ganglion cell tumors: Ganglioneuromas, ganglioneuroblastomas, and neuroblastomas arise from the autonomic nervous system rather than the nerve sheath.
Clinical Presentation
Thymomas usually are an incidental finding, but they can manifest as chest pain, cough, or dyspnea in up to one third of patients.22 Myasthenia gravis, characteristically associated with thymomas, occurs most frequently in women. Thirty percent to 50% of patients with thymomas have myasthenia gravis, whereas 10% to 15% of patients with myasthenia gravis have a thymoma.23 Patients with myasthenia gravis usually have diplopia, ptosis, dysphagia, and fatigue. Ten percent of patients with a thymoma have hypogammaglobulinemia and 5% of patients have pure red cell aplasia. Thymomas are also associated with various autoimmune disorders such as systemic lupus erythematosus, polymyositis, or myocarditis.2 Thymic carcinomas are frequently symptomatic at presentation owing to marked local invasion and involvement of mediastinal structures. Symptoms include chest pain, dyspnea, cough, SVC syndrome, usually due to compression or invasion of the SVC; unlike thymomas, paraneoplastic syndromes are rare. Thymic neuroendocrine tumors are also associated with ectopic secretion of hormones. Up to 50% of affected patients with thymic carcinoid tumors have hormonal abnormalities and up to 35% have Cushing’s syndrome due to tumoral production of adrenocorticotropic hormone (ACTH). Nonfunctioning thymic carcinoids may be seen in association with the multiple endocrine neoplasia syndrome type 1.
Patients with germ cell tumors are often asymptomatic. Large tumors can manifest clinically as cough, dyspnea, chest pain, or pulmonary infection. Rarely, teratomas may erode and rupture into adjacent airway, resulting in expectoration of oily material or hair (tricophtysis). Seminomas can manifest as SVC syndrome in 10% of cases. Beta-human chorionic gonadotropin (β-hCG) and alpha-fetoprotein (AFP) levels are usually normal. However, 7% to 8% of patients with pure seminomas are reported to have elevated serum levels of β-hCG and elevation of serum lactate dehydrogenase (LDH) levels occurs in 80% of patients with advanced seminomas. Importantly, elevation of AFP indicates a nonseminomatous component of the tumor. Most (90%) of patients with nonseminomatous germ cell tumors of the mediastinum are symptomatic at presentation with chest pain, cough, dyspnea, weight loss, and fever. Serologic tumor markers are important in the diagnostic evaluation and 71% of affected patients have elevated levels of AFP and 54% have elevated levels of β-hCG.24 There is an association between malignant nonseminomatous germ cell tumors of the mediastinum and hematologic malignancies. In addition, approximately 20% of cases are associated with Klinefelter’s syndrome (gynecomastia, testicular atrophy, and 47 XXY karyotype).
Nerve sheath tumors are usually asymptomatic. Because most patients with a benign neurogenic tumor are asymptomatic, the development of pain often indicates malignant transformation (malignant neurofibromas, malignant schwannomas, or neurofibrosarcomas). Mediastinal neuroblastomas can manifest clinically due to local mass effect leading to respiratory distress or spinal cord compression. Neuroblastomas and, less frequently, ganglioneuroblastoma and ganglioneuroma can produce metabolically active catecholamines responsible for hypertension, flushing, and watery diarrhea syndrome.25,26 Catecholamine derivatives, such as vanilmandelic acid and homovanilic acid, can be secreted and associated with elevated catecholamine plasma levels and are helpful in establishing the diagnosis.
Key Points Middle and posterior mediastinal masses
• Vascular: Aorta (aneurysm, dissection, and congenital abnormalities), pulmonary artery (aneurysm and pulmonary hypertension), and venous abnormalities (left SVC and azygos/hemiazygos system abnormalities).
• Adenopathy. Infectious (tuberculosis, histoplasmosis, and coccidioidomycosis), sarcoidosis, lymphoma, metastatic disease (head and neck, melanoma, breast, and genitourinary), Castleman’s disease.
• Cysts: Pericardial, esophageal, bronchogenic, meningocele, pancreatic pseudocyst, neurenteric, cystic tumors.
• Esophageal: Megaesophagus, esophageal varices, neoplasms.
• Neurogenic tumors: Nerve sheath (neurofibroma, schwannoma, and malignant tumors of nerve sheath origin), ganglion cell (neuroblastoma, ganglioneuroma, and ganglioneuroblastoma), paraganglia cell (paraganglioma).
• Miscellaneous: Hematoma, abscess, hiatal hernia, congenital hernia.
Staging
In 1999, the World Health Organization (WHO) Consensus Committee published a histologic classification of tumors of the thymus. In this scheme, thymomas are classified on the combined basis of the morphology of the neoplastic epithelial cells and the lymphocyte–to–epithelial cell ratio. This classification has resulted in six separate histologic subtypes of thymomas (types A, AB, B1, B2, B3, and C) (Table 7-1). An update on the WHO classification was published in 2004.27 It retained its classifications of A through B3 thymomas but relegated type C to a separate category of thymic carcinoma and acknowledged that certain types of thymoma do not fall into these categories. The applicability and clinical significance of the WHO histologic classification of thymomas is still being evaluated. It appears that most thymomas can be classified using the WHO criteria, although the clinical and prognostic relevance of this classification has not been conclusively determined. Most thymomas of types A and AB tend to have no local invasion, are completely resectable, and have no recurrence or tumor-related deaths. There is an increasing tendency to local invasion, incomplete resection, and recurrence after resection in the order of types of B1 and B2, type B3, and type C thymomas. In this regard, most type C thymomas are locally invasive, many are incompletely resected and there is a high early relapse rate and poor prognosis. The WHO histologic subtype classification of thymomas has been reported to correlate with the staging classification devised by Masaoka and coworkers. The Masaoka staging system is a pathologic staging system that is based on the presence of capsular invasion and is currently the most widely used to determine therapy.28 In this staging system, the stages are defined as stage I, macroscopically encapsulated and microscopically no capsular invasion; stage II, macroscopic invasion into surrounding fatty tissue of mediastinal pleura or microscopic invasion into capsule; stage III, macroscopic invasion into a neighboring organ; stage IVa, pleural or pericardial dissemination; and stage IVb, lymphogenous or hematogenous metastasis.
Table 7-1 World Health Organization Classification Scheme for Thymic Epithelial Tumors
| TUMOR TYPE | DESCRIPTION |
|---|---|
| A | Medullary |
| AB | Mixed |
| B1 | Lymphocyte rich, predominantly cortical |
| B2 | Cortical |
| B3 | Epithelial (well-differentiated thymic carcinoma) |
| C | Thymic carcinoma |
Neuroblastomas are currently staged according to the International Neuroblastoma Staging System (INSS), which is based on surgical findings as well as lymph node and metastatic involvement. However, new guidelines for a pretreatment staging system have been developed by the International Neuroblastoma Risk Group (INRG); this staging system is being implemented clinically.29 This staging model is based on clinical and imaging features rather than surgical findings. Currently, both of these staging systems have an important role in the determination of appropriate treatment and prediction of outcome in patients with neuroblastoma (Table 7-2).
Table 7-2 Comparison between International Neuroblastoma Staging System and International Neuroblastoma Risk Group Staging System
| INSS | INRGSS |
|---|---|
| Stage 1: Localized tumor with complete gross excision; ± microscopic residual disease; representative ipsilateral lymph node negative for tumor microscopically. | Stage L1: Localized tumor not involving vital structures as defined by IDRFs and confined to one body compartment. |
| Stage 2A: Localized tumor with incomplete gross excision; representative ipsilateral lymph node negative for tumor microscopically. | Stage L2: Locoregional tumor with presence of one or more IDRFs. |
| Stage 2B: Localized tumor with or without complete gross excision; ipsilateral lymph node positive for tumor microscopically; enlarged contralateral lymph nodes should be negative microscopically. | Equals stage L2. |
| Stage 3: Unresectable unilateral tumor infiltrating across the midline; ± regional lymph node involvement; or localized unilateral tumor with contralateral regional lymph node involvement or midline tumor with bilateral extension by infiltration (unresectable) or by lymph node involvement. | Equals stage L2. |
| Stage 4: Any primary tumor with dissemination to distant lymph nodes, bone, bone marrow, liver, skin, or other organs. | Stage M: Distant metastatic disease (except stage MS). Distant lymph node involvement is metastatic disease. Ascites and pleural effusion, even if malignant cells are present, do not constitute metastatic disease unless they are remote from the primary tumor. |
| Stage 4S: Localized primary tumor in infants < 1 yr (localized as in stage 1, 2A, or 2B) with dissemination limited to skin, liver, or bone marrow (<10% malignant cells). | Stage MS: Metastatic disease in children < 547 days (18 mo) of age with metastases confined to skin, liver, and/or bone marrow (<10% malignant cells); MIBG scan must be negative in bone and bone marrow. Primary tumor can be L1 or L2 with no limitations in terms of crossing or infiltration of the midline. |
IDRFs, image-defined risk factors; INRGSS, International Neuroblastoma Risk Group Staging System; INSS, International Neuroblastoma Staging System; MIBG, iodine-123-metaiodobenzylguanidine.
Adapted from Monclair T, Brodeur GM, Ambros PF, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol. 2009;27:298-303.